

Abstract
The different mechanisms employed by proto-oncogenes and tumor suppressors to regulate cell death pathways are strictly linked to their localization. In addition to the canonical control of apoptosis at a transcriptional/nuclear level, intracellular zones are emerging as pivotal sites for the activities of several proapoptotic and antiapoptotic factors. Here, we review the function of the endoplasmic reticulum-mitochondria interface as a primary platform for decoding danger signals as well as a structural accommodation for several regulator or effector proteins.
Keywords: apoptosis, calcium, cancer, endoplasmic reticulum, mitochondria, mitochondria-associated membranes (MAMs), oncogenes, oncosuppressors
Abbreviations
- ER
endoplasmic reticulum
- IMM
inner mitochondrial membrane
- IP3R
inositol 1, 4, 5-trisphosphate receptor
- MAM
membrane-associated membrane
- MCU
mitochondrial Ca2+ uniporter
- mPTP
mitochondrial permeability transition pore
- mTORc2
mechanistic target of rapamycin complex 2
- OMM
outer mitochondrial membrane
- PERK
RNA-dependent protein kinase (PKR)-like ER kinase
- PML
promyelocytic leukemia
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
- ROS
reactive oxygen species
- VDAC
voltage-dependent anion channel
Introduction
Over the past 15 years, alternative subcellular districts have been described as pivotal sites of action for several oncogenes and oncosuppressors. Although the core apoptotic machinery is tightly controlled at the transcriptional level, a series of post-translational mechanisms, such as translocation to different intracellular compartments, phosphorylation events, and protein–protein interactions, represent important aspects of regulation of the apoptotic pathway. Although the nucleus has been identified as the main target of different oncogenes, such as c-myc, and tumor suppressors, such as p53, it is now clear that coordination of the apoptotic process occurs at multiple cellular levels, far removed from the biochemical scheme of one protein–one function. These unconventional pathways probably act in synergy with transcription-dependent pathways, and in some pathological contexts they function not only to support the nuclear mechanisms but can also be considered the primary molecular route.
Among the different non-nuclear activities, mitochondrial signaling is one of the most studied and best characterized. Mitochondria are the major site of localization for oncogenes and oncosuppressors because of their central role as integrators and transducers for proapoptotic signals.1 However, protein targeting to mitochondria generally requires the presence of specific import signals; therefore, a large number of proteins cannot easily enter mitochondria but instead exert their effects in the surrounding zone, especially in the contact areas between the endoplasmic reticulum (ER) and mitochondria.
Membrane-bound organelles exchange metabolic signals and information through the formation of specific membrane contact sites, and the ER-mitochondria interface represents one such connection. The mitochondria and ER join together at multiple contact sites, forming a specified subcellular fraction that is currently termed mitochondria-associated membranes (MAM).2,3 The ER and mitochondria not only physically couple but also establish a tight communication that plays a crucial role in several processes, such as lipid trafficking, calcium (Ca2+)-transfer, inflammation, and apoptosis.2,4,5 Nevertheless, several regulatory factors are shared by the 2 organelles, leading to the establishment of a molecular platform to receive and decode a wide range of inputs, including apoptotic signals. During cellular stress, ER-mitochondria connections lead to the prompt activation of caspase-dependent and caspase-independent cell death effector mechanisms based on the capacity of the 2 organelles to sense and react to a multiple array of danger signals. Thus, the MAM can be conceived as a physical and functional scaffold for the primary response to alterations of cellular homeostasis, and it is therefore logical that oncogenes and tumor suppressors can localize at or move to MAM upon cellular stress.
This review will address the structure of MAM, the main apoptotic-related functions of MAM (with a focus on reactive oxygen species (ROS) and Ca2+ exchange) and the proteins that regulate the cell death pathways through their localization to the ER-mitochondria interface (Table 1).
Table 1.
Summary of the most important oncogenes and oncosuppressors discussed in this review. MAM, mitochondria-associated ER membranes; Mt, mitochondria; PAM, plasma membrane associated membranes; ER, endoplasmic reticulum; Nu, nucleus; Cyt, cytosol; PM, plasma membrane
| Protein | Localization | Functions at MAM | MAM Interactors | References |
|---|---|---|---|---|
| p66Shc | MAM, PAM, Cyt, Mt | Cytosolic adaptor protein involved in cellular response to oxidative stress | Unknown | 24,26 |
| Akt | Cyt, Nu, MAM | Serine/threonine protein kinase. Inhibition of Ca2+ release from ER; antiapoptotic functions | Bad, Bax, HK2, IP3R, PACS2, PTEN, PML, mTORc2 | 33,37,38,40,41 |
| Bcl-2 | Nu, ER, Mt, Cyt, MAM | Induction of Ca2+ leakage from ER; antiapoptotic functions | Bad, Bcl-xL, IP3R | 44,46,47,51 |
| Bcl-xL | Nu, Cyt, Mt | Induction of Ca2+ leakage from ER; antiapoptotic functions | Bad, Bcl-2, IP3R | 57 |
| Bad | Cyt, Mt, ER | Proapoptotic functions | Bcl-2, Bcl-xL | 48,68,69 |
| Bax | Cyt, Mt, ER | Proapoptotic functions | Akt, Bcl-2 | 48,68,69 |
| HK2 | Mt | Glucose phosphorylation; antiapoptotic functions | Akt, VDAC1 | 33,35,36 |
| PTEN | Cyt, Mt, MAM, Nu | Most commonly lost or mutated tumor suppressor in human cancers; negative regulator of Akt, regulation of Ca2+ release via IP3R3; proapoptotic functions | Akt, IP3R, PP2a | 87 |
| PML | MAM, ER, Nu | Implicated in the pathogenesis of leukemia and other cancers; negative regulator of Akt; proapoptotic functions | Akt, IP3R, PP2a | 81 |
| mTORc2 (mTOR complex 2) | ER, Mt, MAM | Serine/threonine protein kinase; Akt activator; control of MAM integrity; regulation of Ca2+ uptake; regulation of mitochondrial bioenergetics; antiapoptotic functions. | Akt, PACS-2 | 41 |
| Sig1-R | MAM/ER | Molecular chaperone stabilizing the conformation of proteins at the MAM; promotes cellular survival; antiapoptotic functions | GRP78/BiP, (IP3R3) | 44,52,53 |
| GRP78/BiP | ER, MT, Cyt, MAM, Nu | ER chaperone - folding and assembly of membrane or secreted proteins; stabilizes IP3R3 at MAM | Sig1-R, CLU | 52,56 |
| CLU | ER, Mt, Cyt | Stress-induced chaperone; antiapoptotic functions | GRP78 Sig1-R | 56 |
| Mcl-1 | MT, Nu, ER, MAM | Induction of Ca2+ leakage from ER; antiapoptotic functions | Bok, IP3R | 58 |
| K-Ras4B | PM, ER, Mt, Cyt | Regulation of Bcl-xL activity; antiapoptotic functions | IP3R, Bcl-xL | 60 |
| H-Ras12v | MAM, PAM, Mt, Cyt, | Regulation of Ca2+ signaling; antiapoptotic functions | Caveolin-1 | 61 |
| K-Ras | MAM, PAM, Mt, Cyt | Inhibition of Ca2+ release from ER and reduction of ER Ca2+levels; antiapoptotic functions | IP3R | 62 |
| vMIA | Mt, MAM | Inhibition of apoptosis | Bax | 63,64 |
| HBx | OMM, Cyt, Nu | Induction of mitochondrial fragmentation and mitophagy; induction of dysfunction of permeability transition pore (PTP) complex | VDAC3 | 65 |
| Enterovirus 2B protein | ER, Golgi-derived vesicles | Regulation of Ca2+ homeostasis; antiapoptotic functions | Unknown | 67 |
| Bok | Golgi, ER, MAM, Cyt | Upstream of Bax and Bak in control of the transmission of ER/MAM-derived apoptotic signals toward mitochondria; proapoptotic activity | Mcl-1, IP3R | 71-74 |
| Ero1-α | MAM, ER | Key controller of oxidative folding and ER redox homeostasis; enriched at MAM and regulates Ca2+ fluxes | IP3R, PAC-1 | 76 |
| Fis1 | MAM, Mt | Formation of a tripartite protein complex with procaspase-8 and Bap31; induction of apoptosis. | Bap31 | 78 |
| Bap31, | MAM, ER, OMM | Formation of a tripartite protein complex with procaspase-8 and Fis1; induction of apoptosis. | Fis1, caspase-8 | 78 |
| PERK | MAM, ER | Involved in folded protein response during ER stress; physically increases contacts between mitochondria and ER | 79 |
MAM structure
It has been demonstrated that, in order to function properly, mitochondria need to communicate with other organelles and intracellular structures. Such communication between the ER and mitochondria can occur at close contact sites between organelles, even without the direct fusion of “interacting” membranes (Fig. 1). Studies on the specific interactions between mitochondria (which form an efficient calcium ion buffer) and the ER (the main intracellular calcium store) were initiated by Copeland and Dalton in the late 1950s. These pioneer studies were performed in cells of the pseudobranch gland of teleost fish and showed that part of the ER exists in association with mitochondria.6 Approximately 10 years later, other groups visualized these contacts in the rat liver and the onion stem by electron microscopy.7,8 Initial evaluation of the extent of the mitochondrial involvement in such interactions gave surprisingly high values, indicating that approximately 80 % of the mitochondrion interacted with the ER. In contrast, later studies showed that only 5–20 % of the mitochondrial surface interacts with the ER. Initially, studies with electron microscopy tomography estimated the distance between the ER and mitochondria as 100 nm, but later studies by Achleitner et al. indicated that this distance varied, and was in the range of only 10–60 nm.9 More recent data further reduced this distance to 10–25 nm, which allows proteins from the ER to associate directly with proteins and lipids present at the outer surface of the mitochondrial membrane.10 Protocols describing the isolation of ER-mitochondrial contacts indicated that these interactions are strong enough to be preserved upon subcellular fractionation. The subcellular fraction that was enriched for the contact sites between mitochondria and ER was named the “mitochondria-associated membrane” (MAM) fraction. A detailed protocol to isolate the MAM fraction was first described by Jean Vance in the early 1990s.11 Since then, the isolation procedure has been improved and adapted to isolate the MAM fraction from different organs, tissues, and various cell lines as well as from yeast.9,11,12 The isolated MAM fraction is composed of membrane shreds from both the ER and outer mitochondrial membrane (OMM) that were in close contact at the time of subcellular fractionation. More recently, the MAM fraction has also been regarded as an intracellular lipid raft of detergent-resistant domains of the ER.13 The contact sites between mitochondria and the ER are dynamically formed as a direct result of a stochastic apposition of ER with mitochondria and are dependent on intracellular signaling. Thus, MAM composition is transient and can be changing at any given time. The variety of roles played by the MAM fraction, as described in the literature, is related to their unique lipid and protein composition. Studies performed in the last decade revealed the molecular components of the MAM fraction, demonstrating that it contains several proteins (more than 75 according to Raturi and Simmen)2 and is crucial for many cellular processes, including protein sorting, inflammation, ER stress, Ca2+ handling, lipid synthesis, trafficking, and apoptosis. However, the localization of some proteins in the MAM fraction and the extent of their enrichment are still under debate because their connection to the MAM fraction is unclear.
Figure 1.
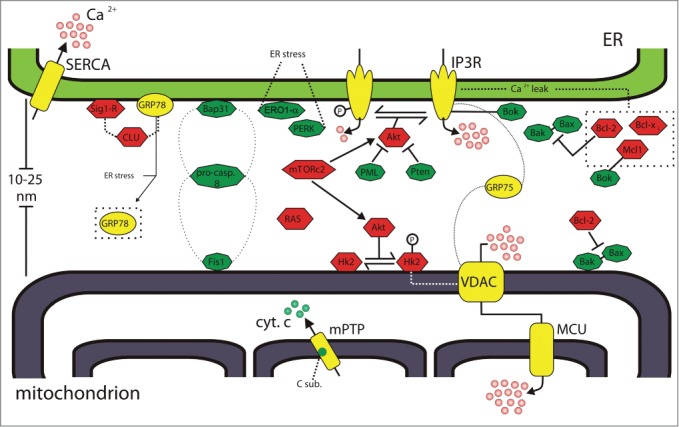
Oncogenes and oncosuppressors at mitochondria-associated membranes. Oncogenes and tumor suppressors acting at the ER–mitochondria interface are shown as red hexagons and green octagons, respectively. Ca2+ players are shown in yellow. GRP75 interacts with IP3R and VDAC to bridge Ca2+ transfer from ER to mitochondria. Bcl-2 counteracts Bax–Bak activities both at mitochondria and ER sides of the MAM. Bok interacts with IP3R and Mcl-1. Akt phosphorylates IP3R, reducing Ca2+ release from the ER. At the mitochondria, Akt promotes the association between Hk-2 and VDAC. Akt activity is positively/negatively regulated by mTORc2, PML, and Pten. Bap31, caspase 8, and Fis1 form a platform to transduce the cell death signals between the ER and mitochondria. GRP78 might translocate from the ER to MAM upon ER stress induction. See text for further details. Bak, Bcl-2-antagonist killer; Bap31, B-cell receptor-associated protein 31; Bax, Bcl-2-associated X protein; Bcl-2, B-cell CLL/lymphoma 2; Bcl-xL, B-cell lymphoma-extra large protein; Bok, Bcl-2-related ovarian killer; CLU, clusterin; C sub, c subunit of mitochondrial ATP synthase; cyt. c, cytochrome c; ER, endoplasmic reticulum; Ero1-α, ER oxidoreductin-1 α; Fis1, Fission 1 homolog; GRP75, glucose regulated protein 75; GRP78, glucose regulated protein 78; Hk2, hexokinase 2; IP3R, inositol 1,4,5 trisphosphate receptor; Mcl-1, myeloid cell leukemia sequence 1; MCU, mitochondrial calcium uniporter; mPTP, mitochondrial permeability transition pore; mTORc2, mechanistic target of rapamycin complex 2; PERK, RNA-dependent protein kinase (PKR)-like ER kinase; PML, promyelocytic leukemia protein; pro-casp. 8, pro-caspase 8; Pten, phosphatase and tensin homolog deleted on chromosome 10; SERCA, sarco/endoplasmatic reticulum Ca2+ ATPase; Sig1-R, Sigma 1 receptor; VDAC, voltage-dependent anion channel.
Originally, the MAM fraction was described as the location of lipid synthesis and trafficking between the ER and mitochondrial membranes based on the presence of long-chain fatty acid-CoA ligase type 4 (FACL4) and phosphatidylserine synthase-1 (PSS-1) enzymes.3 The close apposition of mitochondria to the ER also explains the selective transmission of physiological and pathological Ca2+ and ROS signals directly from the ER to the mitochondria.4 The MAM also contains Ca2+ signaling elements of both organelles, thus supporting the central role of ER/mitochondria crosstalk in signal transduction. Therefore, the ER-mitochondria contact sites can be considered specialized microdomains for the transfer of Ca2+ signals. Ca2+ ions released from the ER by inositol 1,4,5-trisphosphate receptors (IP3Rs) cross the freely permeable OMM through voltage-dependent anion channels (VDACs), reach the inner mitochondrial membrane (IMM), and accumulate in the matrix via the mitochondrial Ca2+ uniporter (MCU) complex. Close apposition between the ER and mitochondria ensures the formation of microdomains at high [Ca2+] that overcome the low apparent Ca2+ affinity of the MCU. Hence, at the "Ca2+ hotspot" stage, the local [Ca2+] is >10 μM, allowing rapid Ca2+ transduction to the matrix despite the low Ca2+ affinity of the uniporter pore (Fig. 1). At the molecular level, the MAM chaperone glucose regulated protein 75 (GRP75) mediates the interaction between the IP3R and VDAC, structurally linking the Ca2+ efflux system at the ER with the channels at the OMM to favor positive regulation of mitochondrial Ca2+ uptake.14 Interestingly, VDAC1, but not VDAC2 and VDAC3, interacts with IP3R, sustaining transmission of the low-amplitude apoptotic Ca2+ signals to mitochondria.15
Although Ca2+ exchange between the ER and mitochondria serves as a regulator of cellular bioenergetics,16 accumulation of Ca2+ can trigger opening of the mitochondrial permeability transition pore (mPTP), leading to release of proapoptotic factors, such as cytochrome c, into the cytosol. The molecular nature of the mPTP is still controversial, but recent evidence suggests the involvement of new structural components in pore formation,17 in particular the c subunit of mitochondrial ATP synthase.18 Conversely, the molecular composition of the MCU complex has been determined19 and its importance in the regulation of cell death pathways has been described in many cellular environments.20,21 Notably, silencing of a core component of the MCU complex, the regulatory subunit mitochondrial calcium uptake 1 (MICU1), exposes the mitochondria to drastic Ca2+ accumulation at basal conditions, produces ROS, and triggers the apoptotic process.22 The ER and mitochondria are 2 of the major sites for ROS production inside the cell.23 Exchange of ROS takes place at the MAM, and this ROS trafficking has a wide relevance in many pathological contexts, especially during ER stress (as discussed under "MAM as a strategic platform for oncosuppressor-dependent cell death"). Therefore, many regulators of the oxidative state of the cell are located at the MAM. p66Shc, a cytosolic adaptor protein involved in a cellular response to oxidative stress, has recently been identified at mitochondria-ER association sites. Moreover, an increasing body of evidence indicates that p66Shc is involved in tumorigenicity. There is a positive relationship between the level of p66Shc and the rate of cell proliferation in prostate cancer cells.24 This relationship is particularly relevant to steroid-induced signaling and elevated levels of ROS, which act as secondary messengers in cancer cells. The upregulation of ROS production by androgen or estrogens is accompanied by an increase in p66Shc, thereby promoting cell proliferation in the aforementioned types of cancer cells. Interestingly, androgen treatment decreases p66Shc phosphorylation at Ser36.24 The association of p66Shc with mitochondrial ROS production has also been repeatedly documented in our previous studies.25 The level of p66Shc in the MAM fraction increases in an age-dependent manner and correlates with mitochondrial ROS production, which has also been found to increase with age.26
Oncogenic function of MAM
As cited in the aforementioned studies, Ca2+ and ROS transfer is the main apoptotic-related function of MAM. By localizing at ER-mitochondria membranes, oncogenes and oncosuppressors can alter this physiological exchange by modifying the cellular response to apoptosis (Fig. 1). This is the case for the serine/threonine kinase Akt. Survival signals involving Akt activation include both the caspase cascade and transcriptional control of apoptosis.27 Upon growth factor stimulation, phosphorylated Akt translocates to the nucleus from the cytoplasm via an activation process. Through a phosphorylation cascade, Akt promotes nuclear exclusion (cytoplasmic retention) of transcription factors of the forkhead family, thereby preventing transcription of the proapoptotic genes Fas ligand, BIM, TRAIL, and TRADD.28 Additionally, phosphorylation of Mdm2 by Akt is necessary for localization of Mdm2 to the nucleus, where it associates with p53 to target its ubiquitination.29 Moreover, Akt-dependent NF-κB nuclear translocation promotes the transcription of antiapoptotic genes, such as BFL1, cIAP1, and cIAP2.27
In addition to its transcriptional activity, Akt has also been physically and functionally linked to both the ER and the mitochondria. Akt-dependent phosphorylation of the proapoptotic BH3-only protein Bad causes its dissociation from the Bcl-2/Bcl-XL complex at the outer mitochondrial membrane, inhibiting its cell death functions.30 Similar to its relationship with Bad, phosphorylation of Bax by Akt results in inhibition of the apoptotic features of Bax, such as oligomerization, insertion into mitochondrial membrane, and the formation of large pores to allow the release of proapoptotic factors.31,32 Akt also phosphorylates hexokinase 2 (HK2) to promote its association with the MAM protein VDAC1. This association not only affects the metabolic state of the cell by increasing efficiency and rate of the glycolytic pathway,33 but also protects cells from apoptosis.34,35 Hexokinase–VDAC1 binding seems to promote the closed state of the channel, preventing Ca2+-dependent opening of the mPTP and release of the proapoptotic protein cytochrome c.36
Furthermore, at the ER side of MAM, Akt phosphorylates all IP3R isoforms,37,38 inhibits Ca2+ release from ER,38,39 and protects cells from apoptosis.37-39 Our group showed that Akt inhibits Ca2+ fluxes and apoptosis in the ER by preferentially phosphorylating the type 3 IP3R (IP3R3).40 IP3R3 is mainly localized to the MAM,2 suggesting a pivotal role of the proto-oncogene Akt at the ER-mitochondria interface. These results provided evidence for the hypothesis of individuation at MAM of both negative regulators of Akt (the tumor suppressors PTEN and PML, see section below) and the Akt activator mechanistic target of rapamycin complex 2 (mTORc2).41 Previous observations suggest that mTORc2 can interact with both the ER and the mitochondria. Indeed, mTORc2 resides at the MAM,41 where it regulates the phosphorylation state of IP3R3 and Ca2+ release from the ER. mTORc2 deficiency, as well as Akt downregulation, causes MAM disruption.41 mTORc2 controls MAM integrity, at least in part, via Akt-dependent phosphorylation of phosphofurin acidic cluster sorting protein 2 (PACS2), as suggested by the observation that PACS2 is a substrate of Akt42 and is required for MAM integrity.43 Moreover, mTORc2 controls mitochondrial functions and physiology in an Akt-dependent manner through HK2 phosphorylation. Thus, mTORc2 localization to MAM can be considered the ideal link to the multiple apoptotic-related functions of Akt, underlining the crucial role of the proto-oncogene at ER–mitochondria contact sites.
As in the case of Akt, strategic positioning at the MAM and regulation of Ca2+ fluxes is shared by other oncoproteins, including the Bcl-2 family members. Bcl-2, the “patriarch” of the family, is highly enriched at the MAM,44 where it functions at both the ER and the mitochondrial side to exert its antiapoptotic function. At the mitochondria, Bcl-2 binds Bax/Bak, preventing their oligomerization and inhibiting Bax/Bak pore formation (for a review, see45). At the ER, Bcl-2 modulates Ca2+ transfer by decreasing net influx into the ER through increased Ca2+ leakage46,47 rather than reduced Ca2+ release. Increased Ca2+ leakage results in reduced Ca2+ transfer to mitochondria and inhibits apoptosis. Nevertheless, Bax/Bak double knockout cells were shown to have a reduced steady state of ER Ca2+, and hence are protected from a variety of apoptotic challenges.48 However, an Akt-like function (i.e., inhibition of ER Ca2+ release), rather than augmented Ca2+ leakage, has also been suggested for Bcl-2,49,50 as the antiapoptotic Ca2+ effect of Bcl-2 might be due to direct interaction of its BH4 domain with IP3R.45,51 In either case, Bcl-2 promotes survival in multiple cellular environments by limiting Ca2+ transfer from the ER to the mitochondria.
Interestingly, Bcl-2 expression, but not stabilization, is significantly regulated by the MAM protein sigma-1 receptor (Sig1-R).44 As a molecular chaperone, Sig1-R interacts with many effectors at MAM, but not with Bcl-2. Conversely, Sig1-R transcriptionally controls the expression of Bcl-2 by regulating the ROS/NF-κB pathway.44 Of note, the Sig1-R-mediated downregulation of bcl-2 mRNA is abolished by ROS scavengers and by the inhibition of NF-κB. Thus, Sig1-R affects cell survival through the regulation of bcl-2 levels, revealing that increased MAM activities during stress conditions can directly impact the response at nuclear level.
Sig-1R is a molecular chaperone that stabilizes the conformation of proteins at the MAM, such as IP3R352 or the ER stress sensor IRE1.53 Sig1-R has been implicated in several human diseases, and one of its most important functions is its robust cellular protective effect. Sig1-R agonists have been shown to promote cellular survival by preventing oxidative stress caused by multiple pathological conditions.54 Moreover, Sig1-R plays a crucial role in the control of ER-mitochondrial interorganelle Ca2+ signaling. Sig1-R at the MAM forms a complex with GRP78 (also known as Bip) to regulate Ca2+ homeostasis between the ER and the mitochondria through IP3R.52 GRP78 is primarily located in the ER lumen, but under ER stress a significant pool of GRP78 is localized in different subcellular compartments, such as the cytosol and mitochondria.55 A recent study reported the association between GRP78 and clusterin (CLU), a stress-induced multifunctional secreted and cytoplasmic molecular chaperone, under ER stress conditions.56 This interaction elicits CLU redistribution on the mitochondria, promoting survival of prostate cancer cells during treatment stress.56
At ER-mitochondria contact sites, a Bcl-2-like activity has been described for other antiapoptotic members of the family. Bcl-xL interacts with IP3Rs and sensitizes them to low IP3 concentrations, thus reducing ER Ca2+ concentrations, stimulating mitochondrial energy, and preserving survival.57 The same molecular mechanism is shared by myeloid cell leukemia sequence 1 (Bcl-2-related) (Mcl-1),58 providing additional evidence for the crucial role of Ca2+ leakage in the prosurvival functions of the antiapoptotic Bcl-2 family subgroup. The localization of Bcl-xL at ER and mitochondria indicates its distinct roles in cell death and Ca2+ homeostasis. Using cell lines derived from ER- and mitochondria-targeted Bcl-xL chimeras that are deficient for Bcl-xL, Li and co-workers showed that ER-targeted Bcl-xL is required to restore Ca2+ homeostasis in knockout cells, whereas mitochondrial localization alone is sufficient to provide protection.59 Recently, the Bcl-xL activity at ER has been linked to a specific form of the Ras oncoprotein, K-Ras4B.60 Phosphorylation of K-Ras4B by protein kinase C promotes its translocation from the plasma membrane to the ER and OMM, which is associated with induction of the cell death pathway. Phospho–K-Ras4B associates with IP3R, limiting the ability of Bcl-xL to sensitize IP3R to the activity of its ligand IP3 and thereby abolishing its typical antiapoptotic role.60
Interestingly, our group has shown that oncogenic H-Ras (H-Ras12v) is localized at both MAM and plasma membrane-associated membranes, suggesting a cooperation between the plasma membrane, the ER, and the mitochondria that is essential for Ca2+ signaling and the maintenance of Ca2+ homeostasis in cancer progression.61 Moreover, oncogenic K-Ras inhibits Ca2+ release from ER, reduces ER Ca2+ levels, and suppresses Ca2+ influx to the mitochondria in colon cancer cell lines.62 Thus, multiple forms of Ras act at the ER–mitochondria interface to manipulate Ca2+ transfer, which in turn contributes to the prosurvival properties of Ras that are associated with the oncogenic phenotype.
In addition to its interaction with cancer-related proteins, the oncogenic functions of MAM extend to some viruses. Human cytomegalovirus encodes multiple antiapoptotic proteins, including viral mitochondrion-localized inhibitor of apoptosis (vMIA). vMIA resides at the MAM,63 where it re-targets proapoptotic Bax to the ER/MAM lipid drafts to induce Bax poly-ubiquitination, proteasome degradation, and consequent inhibition of apoptosis.64
Human hepatitis B virus (HBV) is associated with chronic liver disease and with the development of hepatocellular carcinoma. HBV encodes the regulatory protein HBx, which localizes at the OMM and interacts with the MAM protein VDAC3. HBV/HBx induces mitochondrial fragmentation and mitophagy (the selective autophagic-removal of damaged mitochondria), leading to apoptosis attenuation and most likely viral persistence.65
Lastly, enteroviruses, such as coxsackievirus, poliovirus, and echovirus, confer an antiapoptotic state that not only suppresses the host defense mechanisms but is also protective against cell death induced by pharmacological treatments. The enterovirus 2B protein is localized to the surface of the ER- and Golgi-derived membrane vesicles where viral replication takes place.66 Expression of 2B protein decreases the steady-state Ca2+ levels of both the ER and Golgi, reducing the mitochondrial Ca2+ content and suppressing caspase activation and apoptotic cell death induced by various stimuli.67
MAM as a strategic platform for oncosuppressor-dependent cell death
In recent years it has been demonstrated that many oncosuppressor proteins are localized to the ER and at MAM (Fig. 1), where they modulate different cell death programs.
As described above, different antiapoptotic members of the Bcl-2 family appear to play an important role in modulating Ca2+-dependent apoptotic signals within the mitochondria at the ER and MAM side. In contrast, various proapoptotic members of the same family exert an opposite effect on the ER Ca2+ stores, and thus on the amplitude of Ca2+ signals reaching the key effector during apoptosis, the mitochondria.
In the first phase of Bax protein overexpression, before the catastrophic changes in mitochondrial and ER morphology and other intracellular parameters, there is an increase in ER [Ca2+] levels. This higher ER [Ca2+] content correlates with an increase in mitochondrial Ca2+ loading after activation by stimuli causing the release of Ca2+ from the ER Ca2+ stores.68 As discussed above, these results agree with findings from Bax/Bak knockout embryonic fibroblasts, in which a dramatic reduction in [Ca2+]er, was observed.48 Bcl-2 and Bax/Bak proteins primarily target the IP3R type 1 to affect [Ca2+]er. Indeed, downregulation of IP3R1 counteracted the reduction of [Ca2+]er in cells from Bax/Bak knockout animals,69 which is consistent with the correlation between low expression of IP3R and inhibition of apoptosis.70 These data indicate that Bax/Bak directly counteract the effect of Bcl-2 on Ca2+ signaling.
Recently, another proapoptotic member of the Bcl-2 family, Bcl-2-related ovarian killer (Bok) has recently been described as localized to the ER and the MAM.71 Bok has been shown to selectively interact with Mcl-1 and BFL-1/A1, but not with Bcl-2 or Bcl-xL. Although Bok shares high sequence similarity with Bax and Bak, Bok is not a surrogate of these proteins and is unable to compensate for the combined loss of BAX and BAK in triggering mPTP opening and cell death via apoptosis.72 In contrast, Bok-induced apoptosis is almost blunted in the absence of Bax or Bak, and cells lacking Mcl-1 are significantly more sensitive to Bok-induced apoptosis than control cells.71 These observations suggest that Bok may function upstream of Bax and Bak in the control of the transmission of ER/MAM-derived apoptotic signals toward mitochondria. Furthermore, it has been recently shown that Bok can interact with IP3R channels, affecting IP3R levels by protecting the proteins from proteolytic degradation without modifying their ability to release ER calcium stores.73 However, the role of Bok in regulation of the apoptotic process remains to be elucidated, especially considering recent results obtained in vivo that describe a minimal impact of loss of Bok in mice. Indeed, Bok deficiency in lymphoid and myeloid cells fails to confer any protection against a wide range of apoptotic stimuli and loss of Bok does not accelerate lymphoma development in c-MYC-overexpressing transgenic mice.74
Involvement of the MAM domain is also important in cell death independent of the Bcl-2 family members. Indeed, procaspase-activating compound-1 (PAC-1) (a small molecule that converts procaspase-3 to caspase-3 in cancer cells in vitro and in vivo75) does not require Bax and Bak but is dependent on the engagement of MAM through ER oxidoreductin-1 α (Ero1-α). Ero1-α, a key controller of oxidative folding and ER redox homeostasis, is enriched at MAM and regulates Ca2+ fluxes.76 The efficacy of PAC-1 in activating caspase-3 requires cytochrome c release from the mitochondria, which is induced by mitochondrial Ca2+ overload and an increase in mitochondrial ROS through ER stress that is mediated by p53 upregulated modulator of apoptosis (PUMA) and Ero1-α. ER stress induces ER Ca2+ release that is preferentially transferred into the mitochondria because PAC-1 treatment leads to an increase in the number of MAM via the upregulation of Ero1-α.77
Additionally, the MAM is an important site for the recruitment and processing of procaspase-8 to caspase-8. The proteins participating in this complex are B-cell-receptor-associated protein 31 (Bap31) at the ER and Fission 1 homolog (Fis1) at the mitochondria. The physical interaction between Bap31 and Fis1 provides a tethering force between the ER and the mitochondria, thus facilitating the MAM structure. During the apoptotic program Bap31 is cleaved by caspases. This processing occurs at the MAM with the formation of a tripartite protein complex between procaspase-8, Bap31, and Fis1.78 The key role of the caspase-activated Bap31–Fis1 complex at the MAM is to transduce the cell death signals between the ER and mitochondria. Indeed, Bap31 cleavage (i.e., activation) results in a downstream increase in cytosolic [Ca2+] caused by ER Ca2+ release, leading to mitochondrial Ca2+ uptake.78 This dyshomeostasis of mitochondrial [Ca2+] will trigger PTP opening, causing release of the mitochondrial cofactors into the cytosol to complete the apoptotic process.
Thus, the MAM appears to be a hotspot domain for decoding different intracellular signals of stress. In light of this role, it is not surprising that the p66shc protein (an important ROS sensor involved in apoptosis)26 and the RNA-dependent protein kinase (PKR)-like ER kinase (PERK) (the ER stress sensor of the unfolded protein response) are present at the MAM.79 The PERK protein is particularly enriched at the MAM, where it promotes efficient crosstalk between the ER and the mitochondria based on the transfer of Ca2+ and ROS. PERK appears to be crucial for tethering the ER to the mitochondria and thus for MAM integrity. Indeed, cells deficient in PERK display a fragmented ER structure, causing impairment in the Ca2+ signaling from the ER to the mitochondria that is required for efficient apoptosis. Moreover, destabilization of MAM in the absence of PERK prevents the rapid transfer of ROS from the ER to the mitochondria that is required for the insertion of Bax into the outer mitochondrial membrane and release of cytochrome c from mitochondria via oxidation of the phospholipid cardiolipin.79
In recent years, the increased interest in MAMs, in particular their emerging role in controlling cell death, has led to in-depth analysis of other tumor suppressors that share this intracellular localization.
The promyelocytic leukemia (PML) protein, which is encoded by a tumor suppressor gene implicated in the pathogenesis of leukemia and other cancers, displays both nuclear and cytosolic distribution. At the nucleus, PML forms multiprotein nuclear structures called PML-nuclear bodies (PML-NBs). In the cytosol, PML is associated with endosomes80 and is present at the ER and MAM.81 Studies in knockout mice and cells revealed an essential pleiotropic role for PML in multiple p53-dependent and -independent apoptotic pathways. As a result, PML-null mice and cells are protected from apoptosis triggered by a number of stimuli. We were able to demonstrate that the ER/MAM localization of PML is essential for the apoptotic pathway and orchestrating Ca2+ homeostasis and, eventually, for cell death. In particular, PML modulates IP3R type 3 activities by promoting the formation of a multiprotein complex containing IP3R type 3, Akt, and the protein phosphatase PP2a. In the absence of PML, PP2a is unable to localize to the MAM and thus unable to prevent the IP3R3 phosphorylation mediated by Akt. As described above, hyperphosphorylation of IP3R3 inhibits Ca2+ transfer from the ER to mitochondria, thereby inhibiting the apoptotic process.82
The tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) also localizes to MAM. PTEN is one of the most commonly lost or mutated tumor suppressors in human cancers,83 and germline mutations of PTEN have been found in cancer predisposition syndromes.84 PTEN is a phosphatase that has both lipid and dual-specificity protein phosphatase activity.85 Its growth-attenuating activity has primarily been ascribed to the dephosphorylation of plasma membrane-localized PIP3.86 However, emerging evidence demonstrates that additional PTEN-dependent mechanisms are implicated in tumor suppression. Indeed, PTEN at the MAM regulates ER Ca2+ release via IP3R3 in a protein phosphatase-dependent manner that counteracts Akt phosphorylation of the IP3R3.87 The final result of the action of PTEN on IP3Rs is thus the maintenance of sustained activity of the IP3R3 during the apoptotic stimulation and, consequently, enhanced transfer of Ca2+ from the ER to mitochondria.
Lastly, consistent with the importance of sustained Ca2+ transfer from the ER to the mitochondria for the induction of apoptosis, it has been demonstrated that overexpression of the sarco/endoplasmatic reticulum Ca2+ ATPase (SERCA), a protein that is enriched at the MAM, causes ER [Ca2+] overload, which increases spontaneous88 and induced89 apoptosis, favoring ER-mitochondria Ca2+ transfer and leading to a breakdown of mitochondrial function.4
In conclusion, data emerging from recent literature highlight the MAM as an important hub for the control and integration of apoptosis operated by different oncosuppressors via different signal transduction mechanisms that in many cases share the same Ca2+ signal key effectors. The MAM can be conceived as hotspot sites for Ca2+ homeostasis,5 and any perturbation of the fine regulation of Ca2+ signaling can induce tumor suppressors to switch from a survival role to a cell death mechanism.
Conclusion
The large body of knowledge reviewed here depicts MAM as a regulatory scaffold for the control of cell death. Many factors that localize to the nucleus modulate apoptosis at a transcriptional level but can also reside or translocate to the MAM under stress conditions. Evidence for this role is the reciprocal transmission of different signals between the ER and mitochondria through physical contact. Additionally, MAM may be considered the primary platform for the detection of intracellular danger. We have also reported that manipulation of Ca2+ fluxes represents the primary method of action for several oncogenes and tumor suppressors located at the MAM. IP3R channels, including IP3Rs present at MAM, represent the key targets of different oncogenic and oncosuppressive proteins.90 Indeed, regulation of Ca2+ flux through IP3R is also mediated by ROS; superoxide anions cause oxidation of the IP3R and sensitization of Ca2+ release to promote cytoplasmic Ca2+ oscillations and mitochondrial uptake.91
In addition to Ca2+ input, the MAM provides specialized contact sites for transmitting ROS-mediated signals. In this regard, the role of the MAM protein Sig1-R as an interorganelle signaling modulator has recently been reported,53 providing a new mechanism whereby MAM controls cell fate by conveying the ROS message from the mitochondria to the nucleus.
Characterization of MAM as the main decoder of intracellular danger signals implicates a role for MAM dynamics in several physiopathological scenarios.
Funding Statement
This study was supported by the Italian Association for Cancer Research (AIRC) and local funds from the University of Ferrara to PP and CG; the Italian Ministry of Education, University and Research (COFIN, FIRB, and Futuro in Ricerca) to PP and supported by grant from the Polish Ministry of Science and Higher Education (W100/HFSC/2011) and Grant HFSP RGP0027/2011.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 2012; 13:780-8; PMID:23175281; http://dx.doi.org/ 10.1038/nrm3479 [DOI] [PubMed] [Google Scholar]
- 2. Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta 2013; 1833:213-24; PMID:22575682; http://dx.doi.org/ 10.1016/j.bbamcr.2012.04.013 [DOI] [PubMed] [Google Scholar]
- 3. Kornmann B. The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol 2013; 25:443-8; PMID:23478213; http://dx.doi.org/ 10.1016/j.ceb.2013.02.010 [DOI] [PubMed] [Google Scholar]
- 4. Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 2014; 1837:461-9; PMID:24211533; http://dx.doi.org/ 10.1016/j.bbabio.2013.10.015 [DOI] [PubMed] [Google Scholar]
- 5. Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun Signal 2011; 9:19; PMID:21939514; http://dx.doi.org/ 10.1186/1478-811X-9-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Copeland DE, Dalton AJ. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol 1959; 5:393-6; PMID:13664679; http://dx.doi.org/ 10.1083/jcb.5.3.393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morre DJ, Merritt WD, Lembi CA. Connections between mitochondria and endoplasmic reticulum in rat liver and onion stem. Protoplasma 1971; 73:43-9; PMID:5112775; http://dx.doi.org/ 10.1007/BF01286410 [DOI] [PubMed] [Google Scholar]
- 8. Lewis JA, Tata JR. A rapidly sedimenting fraction of rat liver endoplasmic reticulum. J Cell Sci 1973; 13:447-59; PMID:4357366 [DOI] [PubMed] [Google Scholar]
- 9. Achleitner G, Gaigg B, Krasser A, Kainersdorfer E, Kohlwein SD, Perktold A, Zellnig G, Daum G. Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur J Biochem 1999; 264:545-53; PMID:10491102; http://dx.doi.org/ 10.1046/j.1432-1327.1999.00658.x [DOI] [PubMed] [Google Scholar]
- 10. Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 2006; 174:915-21; PMID:16982799; http://dx.doi.org/ 10.1083/jcb.200604016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vance JE, Stone SJ, Faust JR. Abnormalities in mitochondria-associated membranes and phospholipid biosynthetic enzymes in the mnd/mnd mouse model of neuronal ceroid lipofuscinosis. Biochim Biophys Acta 1997; 1344:286-99; PMID:9059519; http://dx.doi.org/ 10.1016/S0005-2760(96)00153-1 [DOI] [PubMed] [Google Scholar]
- 12. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc 2009; 4:1582-90; PMID:19816421; http://dx.doi.org/ 10.1038/nprot.2009.151 [DOI] [PubMed] [Google Scholar]
- 13. Fujimoto M, Hayashi T, Su TP. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem Biophys Res Commun 2012; 417:635-9; PMID:22185692; http://dx.doi.org/ 10.1016/j.bbrc.2011.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 2006; 175:901-11; PMID:17178908; http://dx.doi.org/ 10.1083/jcb.200608073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ 2012; 19:267-73; PMID:21720385; http://dx.doi.org/ 10.1038/cdd.2011.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270-83; PMID:20655468; http://dx.doi.org/ 10.1016/j.cell.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2014; PMID:24727893; 23343770http://dx.doi.org/10.1038/onc.2014.96 [DOI] [PubMed] [Google Scholar]
- 18. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013; 12:674-83; PMID:23343770; http://dx.doi.org/ 10.4161/cc.23599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol 2014; 592:829-39; PMID:24366263; http://dx.doi.org/ 10.1113/jphysiol.2013.268235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, et al. Downregulation of the Mitochondrial Calcium Uniporter by Cancer-Related miR-25. Curr Biol 2013; 23:58-63; PMID:23246404; http://dx.doi.org/ 10.1016/j.cub.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H, Hardingham GE. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 2013; 4:2034; PMID:23774321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 2012; 151:630-44; PMID:23101630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, et al. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct 2012; 2012:329635; PMID:22175013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rajendran M, Thomes P, Zhang L, Veeramani S, Lin MF. p66Shc–a longevity redox protein in human prostate cancer progression and metastasis : p66Shc in cancer progression and metastasis. Cancer Metastasis Rev 2010; 29:207-22; PMID:20111892; http://dx.doi.org/ 10.1007/s10555-010-9213-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lebiedzinska M, Karkucinska-Wieckowska A, Giorgi C, Karczmarewicz E, Pronicka E, Pinton P, Duszynski J, Pronicki M, Wieckowski MR. Oxidative stress-dependent p66Shc phosphorylation in skin fibroblasts of children with mitochondrial disorders. Biochim Biophys Acta 2010; 1797:952-60; PMID:20226758; http://dx.doi.org/ 10.1016/j.bbabio.2010.03.005 [DOI] [PubMed] [Google Scholar]
- 26. Lebiedzinska M, Duszynski J, Rizzuto R, Pinton P, Wieckowski MR. Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch Biochem Biophys 2009; 486:73-80; PMID:19327338; http://dx.doi.org/ 10.1016/j.abb.2009.03.007 [DOI] [PubMed] [Google Scholar]
- 27. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261-74; PMID:17604717; http://dx.doi.org/ 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96:857-68; PMID:10102273; http://dx.doi.org/ 10.1016/S0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- 29. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A 2001; 98:11598-603; PMID:11504915; http://dx.doi.org/ 10.1073/pnas.181181198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91:231-41; PMID:9346240; http://dx.doi.org/ 10.1016/S0092-8674(00)80405-5 [DOI] [PubMed] [Google Scholar]
- 31. Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, Marrack P, Bratton DL, Henson PM. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 2004; 279:21085-95; PMID:14766748; http://dx.doi.org/ 10.1074/jbc.M400063200 [DOI] [PubMed] [Google Scholar]
- 32. Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene 2001; 20:7779-86; PMID:11753656; http://dx.doi.org/ 10.1038/sj.onc.1204984 [DOI] [PubMed] [Google Scholar]
- 33. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 2001; 15:1406-18; PMID:11390360; http://dx.doi.org/ 10.1101/gad.889901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stiles BL. PI-3-K and AKT: Onto the mitochondria. Adv Drug Deliv Rev 2009; 61:1276-82; PMID:19720099; http://dx.doi.org/ 10.1016/j.addr.2009.07.017 [DOI] [PubMed] [Google Scholar]
- 35. Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem 2013; 288:23798-806; PMID:23836898; http://dx.doi.org/ 10.1074/jbc.M113.482026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J 2004; 377:347-55; PMID:14561215; http://dx.doi.org/ 10.1042/BJ20031465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khan MT, Wagner L, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem 2006; 281:3731-7; PMID:16332683; http://dx.doi.org/ 10.1074/jbc.M509262200 [DOI] [PubMed] [Google Scholar]
- 38. Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Soderberg O, et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A 2008; 105:2427-32; PMID:18250332; http://dx.doi.org/ 10.1073/pnas.0711324105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun 2008; 375:501-5; PMID:18723000; http://dx.doi.org/ 10.1016/j.bbrc.2008.07.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, Pinton P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis 2012; 3:e304; PMID:22552281; http://dx.doi.org/ 10.1038/cddis.2012.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A 2013; 110:12526-34; PMID:23852728; http://dx.doi.org/ 10.1073/pnas.1302455110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aslan JE, You H, Williamson DM, Endig J, Youker RT, Thomas L, Shu H, Du Y, Milewski RL, Brush MH, et al. Akt and 14-3-3 control a PACS-2 homeostatic switch that integrates membrane traffic with TRAIL-induced apoptosis. Mol Cell 2009; 34:497-509; PMID:19481529; http://dx.doi.org/ 10.1016/j.molcel.2009.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 2005; 24:717-29; PMID:15692567; http://dx.doi.org/ 10.1038/sj.emboj.7600559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meunier J, Hayashi T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J Pharmacol Exp Ther 2010; 332:388-97; PMID:19855099; http://dx.doi.org/ 10.1124/jpet.109.160960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Akl H, Vervloessem T, Kiviluoto S, Bittremieux M, Parys JB, De Smedt H, Bultynck G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim Biophys Acta 2014; 1843(10):2240-52; PMID:24768714; 10704437http://dx.doi.org/10.1016/j.bbamcr.2014.04.017 [DOI] [PubMed] [Google Scholar]
- 46. Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 2000; 148:857-62; PMID:10704437; http://dx.doi.org/ 10.1083/jcb.148.5.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N, Krause KH. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci U S A 2000; 97:5723-8; PMID:10823933; http://dx.doi.org/ 10.1073/pnas.97.11.5723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003; 300:135-9; PMID:12624178; http://dx.doi.org/ 10.1126/science.1081208 [DOI] [PubMed] [Google Scholar]
- 49. Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 2004; 166:193-203; PMID:15263017; http://dx.doi.org/ 10.1083/jcb.200309146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H, et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ 2012; 19:295-309; PMID:21818117; http://dx.doi.org/ 10.1038/cdd.2011.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A 2009; 106:14397-402; PMID:19706527; http://dx.doi.org/ 10.1073/pnas.0907555106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007; 131:596-610; PMID:17981125; http://dx.doi.org/ 10.1016/j.cell.2007.08.036 [DOI] [PubMed] [Google Scholar]
- 53. Mori T, Hayashi T, Hayashi E, Su TP. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 2013; 8:e76941; PMID:24204710; http://dx.doi.org/ 10.1371/journal.pone.0076941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci 2010; 31:557-66; PMID:20869780; http://dx.doi.org/ 10.1016/j.tips.2010.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J 2011; 434:181-8; PMID:21309747; http://dx.doi.org/ 10.1042/BJ20101569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li N, Zoubeidi A, Beraldi E, Gleave ME. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene 2013; 32:1933-42; PMID:22689054; http://dx.doi.org/ 10.1038/onc.2012.212 [DOI] [PubMed] [Google Scholar]
- 57. White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 2005; 7:1021-8; PMID:16179951; http://dx.doi.org/ 10.1038/ncb1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem 2010; 285:13678-84; PMID:20189983; http://dx.doi.org/ 10.1074/jbc.M109.096040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eno CO, Eckenrode EF, Olberding KE, Zhao G, White C, Li C. Distinct roles of mitochondria- and ER-localized Bcl-xL in apoptosis resistance and Ca2+ homeostasis. Mol Biol Cell 2012; 23:2605-18; PMID:22573883; http://dx.doi.org/ 10.1091/mbc.E12-02-0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sung PJ, Tsai FD, Vais H, Court H, Yang J, Fehrenbacher N, Foskett JK, Philips MR. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc Natl Acad Sci U S A 2013; 110:20593-8; PMID:24297914; http://dx.doi.org/ 10.1073/pnas.1306431110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rimessi A, Marchi S, Patergnani S, Pinton P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2014; 33:2329-40; PMID:23728347; http://dx.doi.org/ 10.1038/onc.2013.192 [DOI] [PubMed] [Google Scholar]
- 62. Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. J Cell Sci 2014; 127:1607-19; PMID:24522186; http://dx.doi.org/ 10.1242/jcs.141408 [DOI] [PubMed] [Google Scholar]
- 63. Bhuvanendran S, Salka K, Rainey K, Sreetama SC, Williams E, Leeker M, Prasad V, Boyd J, Patterson GH, Jaiswal JK, et al. Superresolution imaging of human cytomegalovirus vMIA localization in sub-mitochondrial compartments. Viruses 2014; 6:1612-36; PMID:24721787; http://dx.doi.org/ 10.3390/v6041612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang A, Hildreth RL, Colberg-Poley AM. Human cytomegalovirus inhibits apoptosis by proteasome-mediated degradation of Bax at endoplasmic reticulum-mitochondrion contacts. J Virol 2013; 87:5657-68; PMID:23487455; http://dx.doi.org/ 10.1128/JVI.00145-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog 2013; 9:e1003722; PMID:24339771; http://dx.doi.org/ 10.1371/journal.ppat.1003722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rust RC, Landmann L, Gosert R, Tang BL, Hong W, Hauri HP, Egger D, Bienz K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J Virol 2001; 75:9808-18; PMID:11559814; http://dx.doi.org/ 10.1128/JVI.75.20.9808-9818.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Campanella M, de Jong AS, Lanke KW, Melchers WJ, Willems PH, Pinton P, Rizzuto R, van Kuppeveld FJ. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J Biol Chem 2004; 279:18440-50; PMID:14976205; http://dx.doi.org/ 10.1074/jbc.M309494200 [DOI] [PubMed] [Google Scholar]
- 68. Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, Rizzuto R. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem 2004; 279:54581-9; PMID:15485871; http://dx.doi.org/ 10.1074/jbc.M409663200 [DOI] [PubMed] [Google Scholar]
- 69. Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A 2005; 102:105-10; PMID:15613488; http://dx.doi.org/ 10.1073/pnas.0408352102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hanson CJ, Bootman MD, Roderick HL. Cell signalling: IP3 receptors channel calcium into cell death. Curr Biol 2004; 14:R933-5; PMID:15530388; http://dx.doi.org/ 10.1016/j.cub.2004.10.019 [DOI] [PubMed] [Google Scholar]
- 71. Echeverry N, Bachmann D, Ke F, Strasser A, Simon HU, Kaufmann T. Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ 2013; 20:785-99; PMID:23429263; http://dx.doi.org/ 10.1038/cdd.2013.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hsu SY, Hsueh AJ. A splicing variant of the Bcl-2 member Bok with a truncated BH3 domain induces apoptosis but does not dimerize with antiapoptotic Bcl-2 proteins in vitro. J Biol Chem 1998; 273:30139-46; PMID:9804769; http://dx.doi.org/ 10.1074/jbc.273.46.30139 [DOI] [PubMed] [Google Scholar]
- 73. Schulman JJ, Wright FA, Kaufmann T, Wojcikiewicz RJ. The Bcl-2 protein family member Bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. J Biol Chem 2013; 288:25340-9; PMID:23884412; http://dx.doi.org/ 10.1074/jbc.M113.496570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N, Bouillet P, Strasser A, Kaufmann T. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ 2012; 19:915-25; PMID:22281706; http://dx.doi.org/ 10.1038/cdd.2011.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon JT, Hwang SK, Jin H, Churchwell MI, Cho MH, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol 2006; 2:543-50; PMID:16936720; http://dx.doi.org/ 10.1038/nchembio814 [DOI] [PubMed] [Google Scholar]
- 76. Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R, Sitia R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid Redox Signal 2012; 16:1077-87; PMID:21854214; http://dx.doi.org/ 10.1089/ars.2011.4004 [DOI] [PubMed] [Google Scholar]
- 77. Seervi M, Sobhan PK, Joseph J, Ann Mathew K, Santhoshkumar TR. ERO1alpha-dependent endoplasmic reticulum-mitochondrial calcium flux contributes to ER stress and mitochondrial permeabilization by procaspase-activating compound-1 (PAC-1). Cell Death Dis 2013; 4:e968; PMID:24357799; http://dx.doi.org/ 10.1038/cddis.2013.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 2011; 30:556-68; PMID:21183955; http://dx.doi.org/ 10.1038/emboj.2010.346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012; 19:1880-91; PMID:22705852; http://dx.doi.org/ 10.1038/cdd.2012.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lin HK, Bergmann S, Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature 2004; 431:205-11; PMID:15356634; http://dx.doi.org/ 10.1038/nature02783 [DOI] [PubMed] [Google Scholar]
- 81. Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010; 330:1247-51; PMID:21030605; http://dx.doi.org/ 10.1126/science.1189157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pinton P, Giorgi C, Pandolfi PP. The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ 2011; 18:1450-6; PMID:21475307; http://dx.doi.org/ 10.1038/cdd.2011.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell 2008; 133:403-14; PMID:18455982; http://dx.doi.org/ 10.1016/j.cell.2008.04.013 [DOI] [PubMed] [Google Scholar]
- 84. Eng C. PTEN: one gene, many syndromes. Hum Mutat 2003; 22:183-98; PMID:12938083; http://dx.doi.org/ 10.1002/humu.10257 [DOI] [PubMed] [Google Scholar]
- 85. Bononi A, Agnoletto C, De Marchi E, Marchi S, Patergnani S, Bonora M, Giorgi C, Missiroli S, Poletti F, Rimessi A, et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res 2011; 2011:329098; PMID:21904669; http://dx.doi.org/ 10.4061/2011/329098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol 2009; 4:127-50; PMID:18767981; http://dx.doi.org/ 10.1146/annurev.pathol.4.110807.092311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, Pandolfi PP, Pinton P. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ 2013; 20:1631-43; PMID:23811847; http://dx.doi.org/ 10.1038/cdd.2013.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ma TS, Mann DL, Lee JH, Gallinghouse GJ. SR compartment calcium and cell apoptosis in SERCA overexpression. Cell Calcium 1999; 26:25-36; PMID:10892568; http://dx.doi.org/ 10.1054/ceca.1999.0049 [DOI] [PubMed] [Google Scholar]
- 89. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J 2001; 20:2690-701; PMID:11387204; http://dx.doi.org/ 10.1093/emboj/20.11.2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Akl H, Bultynck G. Altered Ca(2+) signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta 2013; 1835:180-93; PMID:23232185 [DOI] [PubMed] [Google Scholar]
- 91. Bansaghi S, Golenar T, Madesh M, Csordas G, RamachandraRao S, Sharma K, Yule DI, Joseph SK, Hajnoczky G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J Biol Chem 2014; 289:8170-81; PMID:24469450; http://dx.doi.org/ 10.1074/jbc.M113.504159 [DOI] [PMC free article] [PubMed] [Google Scholar]