

Abstract
The DNA strand exchange protein Rad51 provides a safe mechanism for the repair of DNA breaks using the information of a homologous DNA template. Homologous recombination (HR) also plays a key role in the response to DNA damage that impairs the advance of the replication forks by providing mechanisms to circumvent the lesion and fill in the tracks of single-stranded DNA that are generated during the process of lesion bypass. These activities postpone repair of the blocking lesion to ensure that DNA replication is completed in a timely manner. Experimental evidence generated over the last few years indicates that HR participates in this DNA damage tolerance response together with additional error-free (template switch) and error-prone (translesion synthesis) mechanisms through intricate connections, which are presented here. The choice between repair and tolerance, and the mechanism of tolerance, is critical to avoid increased mutagenesis and/or genome rearrangements, which are both hallmarks of cancer.
Keywords: BRCA2, DNA damage tolerance, DNA repair homologous recombination, Rad18, Rad51, replication
Abbreviations
- BER
base excision repair
- DDT
DNA damage tolerance
- HR
homologous recombination
- HU
hydroxyurea
- HJ
Holliday junction
- NER
nucleotide excision repair
- NHEJ
non-homologous end-joining
- MMS
methylmethane sulfonate
- PCNA
proliferating cell nuclear antigen
- SCJ
sister-chromatid junction
- SSBR
single-strand break repair
- ssDNA
single-stranded DNA
- TLS
translesion synthesis
- UV
ultraviolet
Introduction
Cells have to faithfully replicate their DNA. This task is facilitated by a number of DNA repair mechanisms that “clean” the DNA before replication by specifically dealing with different types of DNA lesions. For example, DNA adducts generated by UV (UV) light are primarily repaired by nucleotide excision repair (NER); alkylated bases and abasic sites are repaired by base excision repair (BER); and DNA breaks are repaired by single-strand break repair (SSBR), homologous recombination (HR), and non-homologous end-joining (NHEJ).1 In many cases, however, the replication forks arrive first and have to face DNA lesions that impair their progression and threaten timely completion of DNA replication. When this occurs, an important decision must be taken: the lesion will either be repaired or bypassed with the repair postponed. DNA damage repair ensures fidelity but can excessively extend S-phase duration and induce genetic instability. In contrast, DNA damage tolerance (DDT) might ensure timely replication, but has to be tightly regulated to prevent excessive mutagenesis and genome rearrangements.2
The first evidence for DDT mechanisms came from studies of Escherichia coli uvr mutants that are defective in NER. When irradiated with UV light these mutants stop replication and accumulate single-stranded DNA (ssDNA; detected by separating pulse-labeled DNA in alkaline sucrose gradients) that is later filled in by a RecA-dependent process. The interpretation of these results was that tracks of ssDNA resulting from the encounter between the replication forks and DNA adducts had accumulated and were subsequently repaired by HR.3 Importantly, although DNA synthesis was restored, the DNA adducts remained unrepaired.4 These observations, and the finding that filling in the gaps could be mutagenic and was therefore performed by error-prone polymerases,5 led to the concept of DDT whereby the lesion that causes the replication block is not repaired but is instead tolerated as a means to complete DNA replication in a timely manner. The first models proposed that the forks were able to bypass or circumvent the lesion, leaving ssDNA gaps behind the fork to be post-replicatively repaired.3 Thus, the term post-replicative repair was coined to define this ssDNA gap filling although this term can be misleading as filling of the gaps might also be coupled to the advance of the fork through the lesion (Fig. 1).
Figure 1.
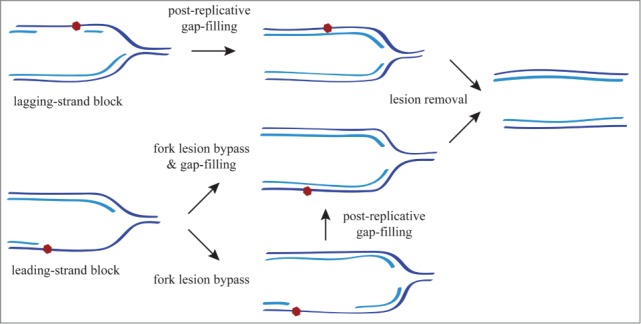
Fork lesion bypass and gap filling after a block of the leading and lagging strand. Blocking the leading strand leads to a transient accumulation of ssDNA through uncoupling of leading and lagging DNA synthesis, whereas blocking the lagging strand is bypassed by priming a new Okazaki fragment. Eukaryotic replication forks can bypass both types of lesions and fill in the ssDNA gaps either during advancement of the fork though the lesion or post-replicatively.
DDT operates in organisms from bacteria to humans. The proteins involved in DDT by HR in the yeast Saccharomyces cerevisiae and in humans, and their corresponding functions, are listed in Table 1. In yeast, Rad51, which is essential in the search for homology and in DNA strand exchange during HR,6 plays a role in DDT appears to be restricted to conditions of high replicative stress. In this article, high and low doses of replicative stress are defined according to whether only the wild-type (low) or both the wild type and the mutants (high) display a discernible biological effect (e.g., cell growth defects).7,8 In higher eukaryotes, however, HR is probably more important for DDT, as inferred from its essential role during DNA replication even in the absence of DNA damaging agents. In any case, the mechanistic contribution of HR during DDT is unclear and in apparent contradiction with the existence of processes that actively prevent HR during S phase in response to replicative stress. Recent molecular and cellular approaches have started to shed some light on the intricate connections between HR and other mechanisms of DDT during the cell cycle. Here, I will first focus on the mechanisms of gap filling in eukaryotes, regardless of whether they are coupled to the fork or operate post-replicatively, and discuss our current knowledge about how the forks can bypass or circumvent a blocking lesion. Next, I will review the mechanisms that regulate DDT during the cell cycle and their impact on genome integrity and cancer.
Table 1.
Proteins involved in DNA damage tolerance through homologous recombinationa
| Yeast | Humans | Functions |
|---|---|---|
| Rad51 | Rad51 | Forms a nucleofilament on ssDNA that is essential for homology search, ssDNA annealing, and strand exchange |
| Rad52 | Rad52; BRCA2 | Mediate the assembly of Rad51 on RPA-coated ssDNA; promote ssDNA annealing and D-loop formation |
| RPA | RPA | ssDNA binding protein; competes with Rad51 for ssDNA binding; modulates DNA damage repair/tolerance and checkpoint activation |
| Rad55/Rad57 | XRCC3/RAD51C/ XRCC2/RAD51B/RAD51D | Rad51 paralogs; counteract Srs2 activity by stabilizing the Rad51 nucleofilament; facilitate Rad51-mediated strand exchange |
| Csm2/Psy3/ Shu1/Shu2/ | RAD51D/SWS1 | Like Rad55/Rad57, the yeast Shu complex counteracts Srs2 activities but its function is restricted to replicative non-DSB DNA lesions |
| Rad54 | Rad54 | Stabilizes Rad51 nucleofilaments and stimulates Rad51-dependent DNA annealing; dissociates Rad51 from dsDNA via its translocase activity; remodels chromatin |
| Mre11/Rad50/Xrs2 | Mre11/Rad50/Nbs1 | Heterotrimeric complex with ATPase and nuclease activities; regulates ssDNA resection; structural role bridging sister chromatids; Nbs1 has an additional role in recruiting Rad18 to UV-induced lesions |
| Sae2 | CtP1; BRCA1 | Initiate DNA resection by counteracting the inhibitory activities of the Ku/DNA-PK complex and Rad9/53BP1; BRCA1 facilitates recruitment of Rad18, HLTF, and Pol η to replicative lesions |
| Exo1 | Exo1 | Structure-specific nuclease involved in processing stalled forks |
| Sgs1/ Top3/Rmi1 | BLM; WRN; SMARCAL1; BLM/TopoIIIα/ Rmi1/Rmi2 | Sgs1, BLM, WRN, and SMARCAL1 are DNA helicases with fork reversal and HJ migration activities; the helicase and topoisomerase activities of Sgs1/BLM and topoisomerase III are required together with Rmi1/Rmi2 for HJ dissolution |
| Ubc9/Mms21 | Ubc9/Mms21 | E2 conjugase (Ubc9) and E3 ligase (Mms21) sumoylation complex that acts in concert with Sgs1/Top3/Rmi1 to resolve HJs, probably by sumoylation of the Smc5-6 complex |
| Smc5–6 | Smc5/Smc6 | Multisubunit complex involved in DSB repair and fork restart by HR and in resolution of MMS-induced SCJs |
| Rad17/Mec3/Ddc1 | Rad9/Rad1/Hus1 | PCNA-like checkpoint clamp; required for full checkpoint activation, and MMS-induced HR in a checkpoint-independent manner |
| Pol δ | Pol δ | Lagging strand synthesis DNA polymerase |
| Pol η | Pol η | TLS polymerase; co-localizes with Rad51 foci after UV irradiation in human cells |
| PCNA | PCNA | Replication processivity factor; loaded by RFC and interacts with DNA polymerases and DNA repair factors via post-transcriptional modifications that control DNA replication and damage tolerance |
| Rad6/ Rad18 | HHR6A; HHR6B/Rad18 | E2 conjugase (Rad6; HHR6A; HHR6B) and E3 ligase (Rad18) ubiquitination complex that monoubiquitinates PCNA to promote TLS; recruited to replicative DNA lesions through the ssDNA binding activity of Rad18 |
| Mms2/Ubc13/ Rad5 | Mms2/Ubc13/HLTF; SHPRH | E2 conjugase (Mms2/Ubc13) and E3 ligase (Rad5, HLTF, SHPRH) ubiquitination complex that extends PCNA K164 ubiquitination with a K63-linked ubiquitin chain to promote template switch; Rad5 and HLTF have also dsDNA translocase activity that promotes fork reversal |
| Mgs1 | WRNIP1; ZRAMB3/AH2 | DNA helicases that bind to ubiquitinated PCNA; Mgs1 destabilizes the interactions between PCNA and Pol δ; ZRAMB3/AH2 has fork reversal and cleavage activity |
| Ubc9/Siz1 | Ubc9/RFC | E2 conjugase (Ubc9) and E3 ligase (Siz1) sumoylation complex that sumoylates PCNA to recruit Srs2; Ubc9 and RFC are sufficient for human PCNA sumoylation in vitro |
| Srs2 | PARI | Sumoylated PCNA interacting DNA helicase that inhibits the salvage HR pathway during S phase by interfering with the formation of ssDNA/Rad51 nucleofilaments and inhibiting DNA repair synthesis |
| Elg1 | ATAD5 | RFC-like clamp loader; interacts with sumoylated PCNA to unload it from chromatin; ATAD5 recruits the ubiquitinating enzyme USP1 |
| Hmo1 | HMGB | Chromatin structural DNA bending protein that binds cruciform structures and promotes Rad51-, Rad5-dependent SCJ formation and prevents function of the salvage HR pathway during S phase |
| Ino80 | Ino80 | ATP-dependent chromatin remodeling complex; facilitates the recruitment of Rad51 and Rad18 to MMS-stressed forks |
aOnly proteins (and their orthologs or functional counterparts) and functions mentioned in the text are shown. Abbreviations: DSB, double-strand break; HJ, Holliday junction; HR, homologous recombination; MMS, methylmethane sulfonate; PCNA, proliferating cell nuclear antigen; RFC, replication factor C; SCJ, sister-chromatid junction; TLS, translesion synthesis.
Rad51 and Rad6/Rad18 Genetic Interactions During DNA Damage Tolerance
Alkaline sucrose gradient analyses in Saccharomyces cerevisiae, in which DDT has been extensively studied, showed early on that eukaryotic cells are also able to fill in ssDNA gaps generated by replicative DNA damage using DDT mechanisms; this led to the elucidation of the first eukaryotic genes involved in gap filling, including the recombination gene RAD52.9-11 Rad52 facilitates loading of Rad51 onto ssDNA filaments, strand invasion, and D-loop formation (Table 1),12 and is essential for most HR events in yeast.13 Supporting a role for HR in DDT, other genes encoding recombination proteins, including RAD51, have subsequently been shown to be required for gap filling.14 However, HR is not central to the replicative DNA damage response in yeast. Indeed, rad52 and rad51 mutants are partially defective in gap filling and are resistant to low doses of agents that damage replicative DNA, such as UV light and the alkylating agent methylmethane sulfonate (MMS).7,8,14
In yeast, the products of RAD6 and RAD18 govern DDT, and accordingly, rad6 and rad18 null mutants are extremely sensitive to UV light or MMS and are completely defective in gap filling.7,8,11 Rad6 and Rad18 form a heterodimer with ubiquitin-conjugating (Rad18) and ubiquitin-ligase (Rad6) activity; additionally, Rad18 has ssDNA binding and ATPase activities.15,16 In response to replicative stress, the Rad6/Rad18 dimer binds to the ssDNA tracks resulting from the encounter of the fork with the blocking lesion and ubiquitinates lysine 164 of the replication processivity factor proliferating cell nuclear antigen (PCNA), a modification that is essential for RAD6-mediated DDT16 (Fig. 2A). Further studies have shown that this pathway is highly conserved from yeast to humans.17
Figure 2.

Mechanisms of gap filling. A replication fork block leads to Rad6/Rad18-dependent PCNA monoubiquitination (A), which targets a specialized polymerase able to insert a correct or incorrect nucleotide opposite the lesion (translesion synthesis) (B). Alternatively, Rad51 and Rad5 can promote ssDNA gap filling by a mechanism that employs the intact sister chromatid to circumvent the lesion through the formation and subsequent dissolution of hemicatenanes (C–H). Hemicatenane formation requires the ssDNA binding complex replication protein A (RPA), the recombination proteins Rad51, Rad52, Rad55, Rad57, the Shu complex, Rad53-dependent phosphorylation of Rad55, Rad54, the nuclease Exo1, the polyubiquitin ligase activity of Rad5, the DNA bending activity of the high mobility group protein Hmo1, the cyclin-dependent kinase Cdc28, and Pol δ, but neither Pol ε nor the TLS polymerases.29,43,44,46,58,84,118 Dissolution of hemicatenanes requires the Sgs1/Top3/Rmi1 complex together with Ubc9/Mms21-dependent sumoylation of the DNA repair Smc5-6 complex.43,119,120 An alternative, Rad5-independent, Rad17/Mec3/Ddc1-dependent salvage HR pathway, which is inhibited in S phase by sumoPCNA-dependent recruitment of Srs2, might be operating through the formation and further resolution of HJs (C–I). See text for more details. * It should be noted that Rad51-strand invasion does not require PCNA monoubiquitination.
Intriguingly, yeast double mutants defective in Rad51 and Rad18 activities are more sensitive than single mutants to UV light and MMS,18 suggesting that HR has RAD6-independent roles in response to replicative DNA damage. This additional function of Rad51 is negatively regulated during S phase by a second modification of PCNA: lysines 164 and 127 are sumoylated by the Ubc9/Siz1 complex both during unperturbed S phase and in response to replicative DNA damage.16 PCNA sumoylation inhibits HR by recruiting the helicase Srs2,18,19 which disrupts the ssDNA/Rad51 nucleofilament and inhibits DNA repair synthesis.20-22 Accordingly, srs2 and PCNA sumoylation-defective mutants (siz1, ubc13, pcna-K164R, K127R) suppress the DNA damage sensitivity of rad6 and rad18, and this suppression is strictly dependent on the HR proteins Rad51, Rad52, Rad54, Rad55, and Rad57.18,19,23 Likewise, in human cells, PCNA is sumoylated at lysine 164, and an Srs2-like protein—PARI—interacts preferentially with sumoylated PCNA to inhibit HR by interfering with the formation of Rad51 nucleofilaments.24 Therefore, Rad51 and Rad6/Rad18 control DDT via intricate and highly regulated functions during the cell cycle.
Mechanisms of ssDNA Gap Filling
As previously mentioned, the accumulation of mutations in response to UV light and alkylating agents suggested the existence of error-prone mechanisms. The search for suppressors of DNA damage-induced mutagenesis, and further biochemical characterization of their corresponding gene products, led to the discovery of specialized polymerases that are able to insert either correct or incorrect bases opposite the lesion.25 In yeast, translesion synthesis (TLS; Fig. 2B) can operate through 2 distinct pathways: an error-prone pathway that involves Rev1 and the polymerase ζ (encoded by REV3 and REV7), and an error-free pathway that involves polymerase η (encoded by RAD30); these polymerases are highly conserved from yeast to humans.2 Importantly, TLS is governed by Rad6/Rad18, which drives filling in of the gaps via monoubiquitination of PCNA.26 This modification, conserved from yeast to humans, facilitates recruitment of the TLS polymerases.25 TLS is not a major repair pathway in yeast,27 and accordingly the sensitivity of a triple rev1 rev3 rad30 (tls) mutant to damaging agents is mild.8 In fact, filling in the UV-induced ssDNA discontinuities is affected only slightly or not at all by the absence of Pol η and Pol ζ , respectively,28 although ssDNA gaps have been detected by different single-molecule analyses in tls yeast mutants 29 and in human cells lacking Pol η.30
Alkaline sucrose gradient studies in yeast have revealed 2 additional groups of genes, defined by RAD52 and RAD5, which additively contribute to gap filling upon UV irradiation.14 Consistent with these findings, double mutants affecting both groups are more sensitive to UV and MMS than single mutants.28 Both pathways require the heterotrimeric complex MRX(N) 14 and operate through error-free mechanisms.31
RAD5 belongs to the same yeast epistasis group of DNA damage sensitivity as UBC13, MMS2, RAD6, and RAD18.31 Yeast Rad5 and its human orthologs helicase-like transcription factor (HLTF) and SNF2 histone linker PHD RING helicase (SHPRH) are proteins with ubiquitin ligase and double-stranded DNA (dsDNA) translocase activities.16,32-34 They recruit the ubiquitin conjugating complex Ubc13/Mms2 to DNA lesions via physical interactions with PCNA and Rad18 and extend Rad6/Rad18-mediated PCNA monoubiquitination at lysine K164 with a K63-linked polyubiquitin chain.17 The function of PCNA polyubiquitination has remained unknown for a long time. Recent work has shown that the yeast helicase Mgs1 is targeted to replicative lesions through an ubiquitin-binding zinc finger domain that physically interacts with the polyubiquitin chain of PCNA. This interaction destabilizes the binding of Pol δ to PCNA, which might facilitate DNA transitions required during the repair process.35 Nonetheless, the main function of PCNA polyubiquitination is unlikely to be linked to Mgs1 because mgs1 null mutants are resistant to DNA damage.36 In human cells, the helicase ZRAMB3/AH2 is also recruited to DNA lesions through its interaction with polyubiquitinated PCNA.37-39 ZRAMB3/AH2 has a number of in vitro activities that suggest different modes of action; specifically, it can promote fork reversal, disrupt D-loop structures, and cut replication forks.37,38
Extensive genetic and molecular analyses in yeast have demonstrated that Rad5 promotes gap filling by a template switch process. Thus, it governs a TLS-independent, error-free process40 and causes sister chromatid exchanges.41 Importantly, Rad5/Mms2/Ubc13 and Rad6/Rad18-dependent sister-chromatid junctions (SCJs) can be molecularly detected in MMS-treated mutants lacking the Sgs1/Top3/Rmi1 complex,42 which is required for SCJ dissolution.43 Indeed, Rad5-dependent SCJs can also be detected in wild-type cells treated with the alkylating agent adozelesin.44 Recently, it has been shown that both yeast Rad5 and human HLTF can promote strand invasion and D-loop formation in vitro, providing the ability to initiate template switching.45
Strikingly, SCJ formation also requires the recombination proteins Rad51, Rad52, Rad55, and Rad57, but not Rad59, which is dispensable for DNA strand exchange.29,42,43,46 In addition, genetic screens aimed at finding new functions that operate in error-free DDT revealed the Rad17/Mec3/Ddc1 (9-1-1) checkpoint complex and its specific DNA loader Rad24.47 Interestingly, this complex is required for functionality of the salvage RAD6-independent HR pathway.47 This HR/9-1-1 pathway is also associated with SCJs, as evidenced by their RAD51-dependent accumulation in sgs1 pcna-K164R, K127R, which is defective in SCJ dissolution and PCNA ubiquitination and sumoylation.42 Together, these results suggest a mechanism in which the sister chromatid provides the template to bypass the lesion through Rad51- and Rad5-mediated strand invasion, DNA synthesis, and SCJ formation; after resolution of these SCJs the lesion persists in a dsDNA molecule and can be removed by oncoming DNA repair mechanisms (Fig. 2C–H). The salvage HR pathway would also contribute to filling the gaps when either replication is completed or inhibition by PCNA sumoylation is removed 42,47 (Fig. 2C–I).
An important and unresolved question is why the salvage HR pathway is repressed during S phase.16,18,19,23 One possibility is that both pathways are initiated by Rad51-mediated strand invasion (Fig. 2C) yet differ in their mechanisms of SCJ formation, acting through a currently undefined Rad5 and PCNA polyubiquitination-dependent process (e.g., extended D-loop by DNA annealing and/or synthesis; Fig. 2D) or a Rad51-mediated strand exchange that is prevented by PCNA sumoylation and Srs2 during S phase (Fig. 2E). Whereas Rad5-mediated D-loop stabilization would lead to the formation of a hemicatenane (Fig. 2F), Rad51-mediated strand exchange would lead to the formation of a Holliday junction (HJ) structure (Fig. 2G). An important distinction between these 2 structures is that the hemicatenanes are “dissolved” by the action of Sgs1/Top3/Rmi1, leading to non-crossovers, whereas HJs can be additionally “resolved” by Mus81/Mms4 and Yen1, leading to non-crossovers and crossovers.48 Crossovers between sister chromatids have no genetic consequences but can be highly deleterious when occurring between DNA sequences located in ectopic or allelic positions (leading to deletions, inversions, translocations, and loss of heterozygosity).49 For this reason, cells restrict the activity of Mus81/Mms4 and Yen1 to late G2/M to resolve SCJs that escape from the dissolution pathway, a strategy that prevents these resolvases from acting on other branched structures as a result of their limited substrate specificity.48 Therefore, upregulation of the salvage HR pathway during S phase would result in an excess of unresolved HJs that could interfere with proper chromosome segregation. SCJs with the properties of hemicatenanes and HJs accumulate in sgs1Δ mutants released into S phase in the presence of MMS; such HJ-like SCJs can be removed by expression of heterologous resolvases.43,50 Interestingly, these resolvases cannot remove early SCJs.50 It is possible that hemicatenanes are initially formed via Rad51 and Rad5-dependent activities and dissolved by Sgs1/Top3/Rmi1 (Fig. 2C–H); in the absence of this complex, the hemicatenanes would be interconverted into HJs and become substrates for resolvases (Fig. 2J–I). The salvage HR pathway would thus be operative in G2/M for unresolved SCJs and in mutants defective for PCNA sumoylation or Srs2 inhibition of the Rad51-mediated strand exchange activity.
The finding of a common pathway for Rad51 and Rad5 raises the question of whether the additive defects in gap filling displayed by mutations affecting the RAD51 and RAD5 groups of genes14 reflect partially overlapping functions or additional and alternative gap filling pathways. Studies with chronic low doses of UV light and MMS suggest that at least one Rad6/Rad18/Rad5-dependent, Rad51-independent DDT pathway may exist because only RAD6, RAD18, and RAD5 are required to deal with low levels of DNA damage in yeast.7,8 Interestingly, evidence for a mechanism involving HR and Rad6/Rad18-specific PCNA monoubiquitination has been reported in human cells, which accumulate DNA repair foci containing both Rad51 and Pol η in response to UV light.51 Indeed, Pol η interacts with Rad51 and can extend DNA synthesis from a 3′-end invading a duplex DNA molecule.52 However, these events could reflect the repair of broken forks, as Pol η is also required for DSB-induced HR.53,54
Replication Fork Lesion Bypass
A growing amount of evidence suggests that gap filling in eukaryotes can occur coupled to the fork and post-replicatively. Data supporting a post-replicative mode of action include the detection by electron microscopy of ssDNA tracks behind the fork in UV- and MMS-treated rad52 and tls mutants in yeast and Xenopus extracts.29,55 In addition, yeast cells in which expression of Rad18, Rad5, Rad17, or TLS polymerases is genetically restricted to G2/M tolerate MMS and UV-induced damage, suggesting that filling of the ssDNA gaps can occur post-replicatively.47,56,57 Likewise, mammalian cells accumulate ssDNA gaps after replication in the presence of UV light, especially in Pol η-defective cells.30 Finally, analysis of Rad52 dynamics in response to MMS in wild-type yeast cells indicates that the recombinational repair of the ssDNA tracks is delayed until G2/M.58 In fact, as mentioned before, the negative regulation of the salvage HR pathway by PCNA sumoylation suggests that at least this pathway operates post-replicatively.
Whereas ssDNA gap filling at the fork directly provides a mechanism for the fork to bypass a blocking lesion, post-replicative repair requires additional mechanisms by which the replication fork either bypasses or circumvents the lesion. This point is intimately related to the position of the blocking lesion relative to the advancing replicative polymerase. Studies in bacteria have shown that a lesion in the leading strand blocks the replication fork, which accumulates ssDNA by transient lagging-strand synthesis, whereas a lesion on the lagging strand inhibits synthesis of the corresponding Okazaki fragment but the fork can bypass this by priming a new DNA fragment 59-61 (Fig. 1).
Indeed, lesions that impair the advance of the leading strand, such as photoproducts or alkylated bases, and the lack of Pol32, which is required for lagging-strand synthesis, both induce PCNA mono- and polyubiquitination, suggesting that gap filling operates in both strands.56 Electron microscopy analyses in yeast and Xenopus extracts have shown that replicative DNA adducts uncouple DNA synthesis and unwinding—thereby generating ssDNA tracks at the fork—in only one of the 2 sisters, yet lead to the accumulation of ssDNA gaps behind the fork in both sisters.29,55 Therefore, eukaryotic replication forks are able to bypass a blocking lesion in both the leading and the lagging strands, even though the leading strand is transitorily stalled and uncoupled from the lagging strand.
Mammalian cells possess a TLS polymerase, PrimPol, that has also a primase activity able to prime new DNA synthesis with dNTPs downstream of a blocking lesion62 (Fig. 3A). This primase activity is employed to reinitiate DNA synthesis after UV irradiation and, accordingly, its downregulation reduces fork progression, underscoring the relevance of this mechanism for replication fork lesion bypass in human cells.63 Yeast lacks a homolog of PrimPol, and it is unknown whether the replicative primase can fulfil this function. On the other hand, tls mutations in yeast do not increase UV-induced fork uncoupling29 or affect fork progression,64 and the expression of TLS polymerases can be restricted to G2/M without affecting DDT,56 suggesting that TLS in yeast operates exclusively at ssDNA gaps left behind the fork. Whereas a putative role for Pol η in replication fork lesion bypass after UV irradiation in human cells remains controversial,30,65 the TLS polymerase Rev1 is required in avian DT40 cells for fork progression through damaged DNA. However, this unexpectedly involves a Rad18- and PCNA monoubiquitination-independent mechanism66 (Fig. 3B).
Figure 3.

Mechanisms of replication fork lesion bypass. Replication forks can bypass a blocking lesion by PrimPol-mediated DNA synthesis downstream of the lesion (A), a switch of the replicative polymerase for a TLS polymerase (B), Rad5 and Rad51-mediated SCJs (B), and fork reversal followed by either Rad51-mediated strand invasion and HJ formation downstream of the blocking lesion (D–E) or by DNA synthesis and fork regression (D–F). Note that Rad51-mediated strand invasion (step E) would require “chicken foot” processing to generate a 3′-ssDNA overhang. See text for details.
A highly conserved and essential mechanism of replication fork lesion bypass relies on HR. Thus, vertebrate cells without Rad51 are not viable as a consequence of defective replication and repair of spontaneous DNA lesions.67,68 After DNA damage, replication is slowed down by the activity of Rad51 and its paralog XRCC3, indicating that HR modulates fork progression through damaged DNA.69 In yeast, HR is not essential for unperturbed replication but appears to be the major mechanism by which the fork bypasses lesions at high levels of DNA damage. Different molecular and cellular approaches have clearly demonstrated that replication through alkylated DNA requires Rad52 and Rad51.44,58,70,71 In fact, Rad52 and Rad51 travel with the fork under unperturbed conditions, and this interaction is not enhanced by replicative DNA damage.55,58 More importantly, yeast cells in which Rad52 expression is restricted to G2/M are defective in DNA replication and in the repair of MMS-induced DNA lesions, indicating that Rad52-dependent loading of Rad51 during S phase is a prerequisite for their post-replicative functions and, further, that replicative and repair HR activities are mechanistically connected.58 A separation of replicative and repair functions has also been shown in mammalian cells, which confront replicative stress by distinct replication fork restart and DNA repair Rad51 mechanisms.72
How Rad51 facilitates replication past DNA lesions is unclear, but the use of adozelesin, an alkylating agent that causes a detectable block of the forks in yeast, has shown that this block is associated with an accumulation of Rad51- and Rad5-dependent SCJs, and that recovery of this block depends on Rad52 and Rad5.44 This suggests that the Rad51/Rad5 pathway (Fig. 2C–H) might be operating at the fork to concomitantly promote fork lesion bypass and gap filling (Fig. 3C).
It is important to note, however, that the role of PCNA ubiquitination in replication fork lesion bypass remains controversial. Thus, although Rad18 and Rad5/HLTF are both required for efficient replication through damaged DNA in yeast and humans,34,70,73 restriction of Rad18 or Rad5 expression to G2/M in yeast does not prevent bulk chromosomal replication in the presence of MMS or UV light.56,57
Notably, the dsDNA translocase activity of yeast Rad5 and human HLTF is able to form a “chicken foot” structure in vitro by fork reversal (Fig. 3D).33,34 This structure, first proposed from mammalian blocked forks,74 might facilitate replication restart downstream of the blocking lesion by Rad51-mediated strand invasion and HJ formation (Fig. 3E) or, alternatively, lesion bypass by DNA synthesis and fork regression (Fig. 3F). However, the helicase domain of Rad5/HLTF, which is required for its dsDNA translocase activity, appears to be dispensable for DDT in vivo,75 suggesting that fork reversal either does not occur, is mediated by a different protein, and/or can be bypassed by an alternate mechanism in the absence of Rad5. In fact, fork reversal can be promoted by the helicase activity of RecQ and SMARCAL1,76 the strand exchange activity of Rad51,77 and even by topological tension.78
Rad51 and the mediator protein BRCA2 are also required to stabilize the stalled forks after dNTP depletion with hydroxyurea (HU) in mammalian cells.79,80 Interestingly, BRCA2 and Rad51 protect HU-stalled forks from degradation by the nuclease Mre11 through a repair-independent mechanism,80 further reinforcing the separation of replicative and repair functions of Rad51 in DDT. Likewise, yeast Rad51 is required for the stability of stalled replication forks.81 Whether this function is associated with the formation of SCJs is unclear because X-shaped structures have not been detected in response to HU,43 but Rad51-mediated fork stabilization also requires the helicase activity of Sgs1 and Top3.81 Thus, it is possible that SCJs need to be rapidly processed after fork stalling by HU. In accordance with this, PCNA polyubiquitination, which is required for SCJs formation,42,44 is also induced by HU.56 In any case, SCJ dissolution is not required for replication fork advance in the presence of replicative stress.43,56 In mammalian cells, Rad51-mediated restart after HU treatment also requires BRCA1 and CtIP, which regulate DNA resection.82 Interestingly, BRCA1 has recently been shown to directly regulate DDT at stalled replication forks by promoting PCNA ubiquitination and physically interacting with Rad18, HLTF, Pol η, and Rev1.83
Regulation of DNA Damage Tolerance
As implicated in the discussion about the mechanisms of DDT, the homotrimeric PCNA sliding clamp plays a decisive role in the choice of mechanism. Whereas the levels of mono- and polyubiquitinated PCNA promote TLS and error-free template switching, respectively,16,26 PCNA sumoylation controls the salvage pathway of HR during S phase by mechanisms that involve the antirecombinogenic activities of Srs216,18,19 as well as the DNA bending activity of Hmo1.84 The relevance of PCNA modifications is highlighted in mammalian cells, in which a higher level of DDT regulation is in part achieved through multiple factors (e.g., p21, p53, Claspin) acting on PCNA ubiquitination.85 The levels of PCNA modification are additionally controlled by the clamp loader Elg1, which binds to and unloads sumoylated PCNA from chromatin in yeast86,87 and targets the PCNA deubiquitinating enzyme USP1 to replicative DNA lesions in humans.88 Chromatin is also involved in this regulation, and the chromatin remodeler Ino80 is required to recruit Rad18 and Rad51 to promote replication fork progression through alkylated DNA.89 Mutations in these regulators lead to unscheduled events that are often associated with severe defects in genome integrity and cancer predisposition (see below). Therefore, PCNA acts as an exquisitely regulated molecular switch controlling different DDT pathways, both in unperturbed replication and in response to replicative DNA damage.
DNA damage sensitivity and molecular assays in yeast have shown that the error-free mechanisms are the major DDT pathways both under low and high replicative stress,7,8,11,27,41 although TLS pathways can also efficiently deal with low doses of DNA damage.7 Intriguingly, the loss of viability of rad18 mutants under both DNA damage conditions can be rescued by reactivating the expression of Rad18 in G2/M in the absence of Rad5, but not of Rev3, indicating that Rad5 requires Rad18 during S phase to tolerate DNA damage.7,57 This, together with the facts that lack of Mms2 (but not of Rev3) delays G2,7 that PCNA mono- and polyubiquitination is initially detected in S phase,56,57 and that the checkpoint is activated during S phase in cells that either lack Rad5 or express it in G2,56 strongly suggests that the RAD5 error-free pathway operates preferentially during S phase, regardless of whether its activity is at or behind the fork. In this scenario, TLS would normally be confined to G2 to deal with any lesions that remained unrepaired. Consistent with this model from data in yeast, the levels of Rev1 display an expression peak during G2/M that is approximately 50-fold higher than that in G1 and S phase and is only minimally affected by DNA damage.90
As previously mentioned, a number of recent results from different eukaryotic models strongly suggest that the replicative and repair activities of the recombination machinery are separated in the response to replicative stress.55,58,72,80 In yeast, this separation of functions is regulated by the replicative checkpoint, which inhibits the formation of HR repair centers during S phase despite the fact that Rad51 and Rad52 remain bound to replicative DNA lesions.58
The type and amount of DNA lesions are also critical determinants in the choice of the DDT mechanism. Thus, SHPRH and HLTF prevent mutations induced by MMS and UV light, respectively, by interacting with Rad18 and promoting recruitment of the most appropriate TLS polymerases.91 Likewise, yeast Rad5, but not Ubc13-dependent PCNA polyubiquitination, is required for UV-induced mutagenesis,92 indicating the existence of additional and conserved intricate connections between the error-free and error-prone pathways.
The importance of the load of DNA lesions has already been anticipated by the differential genetic requirements displayed by yeast in response to an acute high dose and a chronic low-dose treatment. Chronic low doses of DNA damage do not activate the DNA damage checkpoints; indeed, checkpoints are not required for viability under these conditions and become activated and arrest cells in G2/M only in the absence of Rad18 as a consequence of an accumulation of ssDNA that is left unrepaired.7,8 In contrast, the checkpoints are activated even in wild-type cells after an acute high-dose treatment.78 Thus, checkpoint activation by unrepaired ssDNA gaps provides the time required for repair and tolerance mechanisms to deal with the lesions before the cell progresses into mitosis; accordingly, checkpoint defective cells are sensitive to replicative DNA lesions.93
Notably, checkpoint null mutants in yeast are proficient in TLS but partially defective in gap filling,93,94 yet it is unclear whether this effect is related to the roles of the replicative checkpoint in maintaining the stability of stressed replication forks or in the regulation of DDT factors.78 For example, phosphorylation of Rad51 on Ser192 by the sensor checkpoint kinase Mec1 is essential for growth in the presence of MMS.95 In any case, and separate from coordinating repair with cell cycle progression, the checkpoint machinery appears to modulate DDT in response to the load of DNA damage, as suggested by the observation that a partial reduction in the activity of the yeast checkpoint effector Rad53 increases tolerance to MMS by augmenting the amount of Rev1 bound to chromatin and TLS activity.96 This finding is somehow unexpected because, rather than supporting TLS as a backup mechanism for situations of high stress, it suggests that TLS might be permitted only under conditions of low DNA damage. As proposed for bacteria,97 the logic behind this strategy could be modulation of TLS to ensure a certain level of mutagenesis for genetic diversity while preventing a deleterious level of mutations when the number of DNA lesions is excessive.
DNA Damage Tolerance Versus Repair
In yeast, DNA damage repair and DDT mechanisms are both essential when the load of DNA lesions is high.1 However, NER-defective cells can efficiently replicate plasmids carrying a photoproduct.41 In addition, NER and BER repair pathways are dispensable when cells are exposed to chronic low doses of UV light or MMS, whereas Rad6, Rad18, and Rad5 are essential for viability.7,8 Importantly, this loss of viability is not due to a defect in the removal of the blocking lesions, as would be expected for a DDT mechanism.8 Therefore, when yeast forks face DNA blocking lesions, the decision is biased toward tolerating the damage and completing replication. Similar experiments in human cells are lacking, but the high sensitivity to sunlight (chronic low doses) displayed by cells from NER-defective patients98 and the fact that NER is active in the S phase of human, but not yeast, cells99 highlight the importance of DNA damage repair during replication in mammalian cells.
DNA Damage Tolerance, Homologous Recombination, and Cancer
Recent experimental findings suggest that oncogenes induce replicative stress in precancerous cells, which thereby activate replicative checkpoints and DNA repair/tolerance mechanisms that act as a barrier to prevent the proliferation of cells with high levels of genetic instability. Mutations that impair these mechanisms promote cell proliferation and increase genomic instability and, consequently, are selected during tumorigenesis.100 This scenario highlights the importance of HR and DDT in cancer.
The isolation of Pol η as the protein responsible for xeroderma pigmentosum variant (XPV), an inherited genetic disorder that is associated with increased incidence of skin cancers, provided the first evidence for a direct connection between TLS, mutagenesis, and cancer.101 Interestingly, the tumor suppressor genes p53 and p21 negatively control TLS, thus reducing excessive mutagenesis.102 Likewise, the Rad5-mediated pathway appears to play an important role in preventing cancer, as suggested by the high incidence of HLTF gene silencing in human colon cancer 103 and the formation of intestinal cancers in Htlf-deficient mice.104 In fact, a role for Rad5 in tumor suppression is supported by the observation that yeast Rad5 and human HLTF and SHPRH prevent chromosomal rearrangements and mutagenesis.32,105,106 The importance of maintaining strict control of the state of PCNA modifications is also exemplified by the genomic instability and high incidence of tumors in mice that are haploinsufficient for Atad5 (the mouse Elg1 homolog).107
However, HR displays the most critical connections with cancer development. HR plays an active role in preventing tumorigenesis, as can be inferred from its essential functions in replication and repair during replicative stress and as evidenced by the number of tumor suppressor genes that are directly involved in HR.108 Importantly, HR can also be a source of genetic instability, which fuels tumor progression. Thus, HR is highly mutagenic compared to normal replication (1,000-fold more mutagenic)109,110 and can lead to loss of heterozygosity and genome rearrangements when it uses allelic and ectopic DNA sequences to repair the lesions.13 In addition, the accumulation of strand exchange intermediates during DDT can in turn have deleterious effects on genome integrity if they are not tightly controlled. Accordingly, HR intermediates are inhibited during S phase by different mechanisms, such as PCNA sumoylation-dependent recruitment of the anti-recombinogenic helicase Srs2,18,19 checkpoint-dependent regulation of the fork reversal activity of the helicase SMARCAL1 that prevents fork cleavage and unscheduled recombinational repair,111 and cell cycle-regulated recruitment of proteins involved in SCJ dissolution that prevent crossovers and genetic instability.112 Interestingly, the tumor suppressor protein p53, considered to be the guardian of the genome, inhibits replication-associated HR.113 p53 inhibits human Rad51/ssDNA formation, strand exchange, HJ branch migration, and Rad51-mediated fork reversal, probably by direct contact with Rad51.77 Therefore, cells have to tightly control HR to allow its essential functions in DNA replication while simultaneously preventing the deleterious effects of unscheduled recombination events. In yeast, activation of the replicative checkpoints inhibits the formation of recombination centers during S phase.58,71,114-116 I have recently proposed that this inhibition might provide a mechanism to coordinate replicative and repair activities and to prevent genetic instability, since the assembly of repair factories at stressed replication forks might interfere with proper DNA replication and promote fork restart by unscheduled and mutagenic break-induced replication and fork stalling and template switching events using the homology provided by ectopic DNA sequences.117
We have only just started to glimpse the intricate mechanistic connections underlying DDT in eukaryotes, and in particular the critical roles of Rad51 and other recombination proteins such as the tumor suppressor genes BRCA1 and BRCA2. A better understanding of the different DDT pathways will require us to differentially address parameters such as the load of DNA damage, its control by the checkpoints, and DNA lesion specificity. A particular challenge will be deciphering the mechanisms that regulate the replicative and repair activities of HR during the cell cycle, a separation of functions that might also be shared by other DDT mechanisms and which seems to be critical for protection of the genome and prevention of cancer. In addition to the basic and evolutionarily conserved mechanisms ruling DDT addressed in this review, we need to gain a deeper insight into the factors and mechanisms that specifically control the complex DNA damage response in human cells and the genetic consequences of their misregulation.
Funding Statement
The research was funded by the Spanish Ministry of Economy and Competitiveness (BFU2012-38171).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I thank Pedro San-Segundo, Fernando Monje-Casas and Pablo Huertas for critically reading the manuscript.
References
- 1. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedberg EC. Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol 2005; 6:943-53.; PMID:16341080; http://dx.doi.org/ 10.1038/nrm1781 [DOI] [PubMed] [Google Scholar]
- 3. Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of escherichia coli following ultraviolet irradiation. J Mol Biol 1968; 31:291-304; PMID:4865486; http://dx.doi.org/ 10.1016/0022-2836(68)90445-2 [DOI] [PubMed] [Google Scholar]
- 4. Ganesan AK. Persistenceof pyrimidine dimers during post-replication repair in ultraviolet light-irradiated escherichia coli K12. J Mol Biol 1974; 87:103-19; PMID:4610149; http://dx.doi.org/ 10.1016/0022-2836(74)90563-4 [DOI] [PubMed] [Google Scholar]
- 5. Radman M. Molecular and Environmental Aspects of Mutagenesis (eds Prakash L, Sherman F, Miller MW, Tabor H.). 1974; 128-142 [Google Scholar]
- 6. San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229-57.; PMID:18275380; http://dx.doi.org/ 10.1146/annurev.biochem.77.061306.125255 [DOI] [PubMed] [Google Scholar]
- 7. Huang D, Piening BD, Paulovich AG. The preference for error-free or error-prone postreplication repair in Saccharomyces cerevisiae exposed to low-dose methyl methanesulfonate is cell cycle dependent. Mol Cell Biol 2013; 33:1515-27; PMID:23382077; http://dx.doi.org/ 10.1128/MCB.01392-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hishida T, Kubota Y, Carr AM, Iwasaki H. RAD6-RAD18-RAD5-pathway-dependent tolerance to chronic low-dose ultraviolet light. Nature 2009; 457:612-5; PMID:19079240; http://dx.doi.org/ 10.1038/nature07580 [DOI] [PubMed] [Google Scholar]
- 9. Resnick MA, Boyce J, Cox B. Postreplication repair in Saccharomyces cerevisiae. J Bacteriol 1981; 146:285-90; PMID:7012117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jachymczyk WJ, Chlebowicz E, Swietlinska Z, Zuk J. Alkaline sucrose sedimentation studies of MMS-induced DNA single-strand breakage and rejoining in the wild type and in UV-sensitive mutants of Saccharomyces cerevisiae. Mutat Res 1977; 43:1-10; PMID:194147; http://dx.doi.org/ 10.1016/0027-5107(77)90126-9 [DOI] [PubMed] [Google Scholar]
- 11. Prakash L. Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol Gen Genet 1981; 184:471-8; PMID:7038396; http://dx.doi.org/ 10.1007/BF00352525 [DOI] [PubMed] [Google Scholar]
- 12. Mortensen UH, Lisby M, Rothstein R. Rad52. Curr Biol 2009; 19:676-7; PMID:19706272; http://dx.doi.org/ 10.1016/j.cub.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 13. Prado F, Cortés-Ledesma F, Huertas P, Aguilera A. Mitotic recombination in Saccharomyces cerevisiae. Curr Genet 2003; 42:185-98; PMID:12589470 [DOI] [PubMed] [Google Scholar]
- 14. Gangavarapu V, Prakash S, Prakash L. Requirement of RAD52 group genes for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol 2007; 27:7758-64; PMID:17785441; http://dx.doi.org/ 10.1128/MCB.01331-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bailly V, Lauder S, Prakash S, Prakash L. Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J Biol Chem 1997; 272:23360-5; PMID:9287349; http://dx.doi.org/ 10.1074/jbc.272.37.23360 [DOI] [PubMed] [Google Scholar]
- 16. Hoege C, Pfander B, Moldovan G-L, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002; 419:135-41; PMID:12226657; http://dx.doi.org/ 10.1038/nature00991 [DOI] [PubMed] [Google Scholar]
- 17. Unk I, Hajdu I, Blastyák A, Haracska L. Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair 2010; 9:257-67; PMID:20096653; http://dx.doi.org/ 10.1016/j.dnarep.2009.12.013 [DOI] [PubMed] [Google Scholar]
- 18. Pfander B, Moldovan G-L, Sacher M, Hoege C, Jentsch S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005; 436:428-33; PMID:15931174 [DOI] [PubMed] [Google Scholar]
- 19. Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 2005; 19:123-33; PMID:15989970; http://dx.doi.org/ 10.1016/j.molcel.2005.06.001 [DOI] [PubMed] [Google Scholar]
- 20. Krejci L, van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 2003; 423:305-9; PMID:12748644; http://dx.doi.org/ 10.1038/nature01577 [DOI] [PubMed] [Google Scholar]
- 21. Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 2003; 423:309-12; PMID:12748645; http://dx.doi.org/ 10.1038/nature01585 [DOI] [PubMed] [Google Scholar]
- 22. Burkovics P, Sebesta M, Sisakova A, Plault N, Szukacsov V, Robert T, Pintér L, Marini V, Kolesar P, Haracska L, et al. . Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J 2013; 32:742-55; PMID:23395907; http://dx.doi.org/ 10.1038/emboj.2013.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schiestl RH, Prakash S, Prakash L. The SRS2 suppressor of rad6 mutations of Saccharomyces cerevisiae acts by channeling DNA lesions into the RAD52 DNA repair pathway. Genetics 1990; 124:817-31.; PMID:2182387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moldovan G-L, Dejsuphong D, Petalcorin MIR, Hofmann K, Takeda S, Boulton SJ, D’Andrea AD. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 2012; 45:75-86; PMID:22153967; http://dx.doi.org/ 10.1016/j.molcel.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nature 2012; 13:141-52; PMID:22358330; http://dx.doi.org/ 10.1038/nrm3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003; 425:188-91; PMID:12968183; http://dx.doi.org/ 10.1038/nature01965 [DOI] [PubMed] [Google Scholar]
- 27. Baynton K, Bresson-Roy A, Fuchs RP. Analysis of damage tolerance pathways in Saccharomyces cerevisiae: a requirement for Rev3 DNA polymerase in translesion synthesis. Mol Cell Biol 1998; 18:960-6; PMID:9447993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Torres-Ramos CA, Prakash S, Prakash L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol 2002; 22:2419-26; PMID:11884624; http://dx.doi.org/ 10.1128/MCB.22.7.2419-2426.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 2006; 21:15-27; PMID:16387650; http://dx.doi.org/ 10.1016/j.molcel.2005.11.015 [DOI] [PubMed] [Google Scholar]
- 30. Elvers I, Johansson F, Groth P, Erixon K, Helleday T. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res 2011; 39:7049-57; PMID:21646340; http://dx.doi.org/ 10.1093/nar/gkr420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Broomfield S, Hryciw T, Xiao W. DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutat Res 2001; 486:167-84; PMID:11459630; http://dx.doi.org/ 10.1016/S0921-8777(01)00091-X [DOI] [PubMed] [Google Scholar]
- 32. Motegi A, Liaw H-J, Lee K-Y, Roest HP, Maas A, Wu X, Moinova H, Markowitz SD, Ding H, Hoeijmakers JHJ, et al. . Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci USA 2008; 105:12411-6; PMID:18719106; http://dx.doi.org/ 10.1073/pnas.0805685105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blastyák A, Pintér L, Unk I, Prakash L, Prakash S, Haracska L. Yeast Rad5 protein required for postreplication repair Has a DNA helicase activity specific for replication fork regression. Mol Cell 2007; 28:167-75; http://dx.doi.org/ 10.1016/j.molcel.2007.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blastyak A, Hajdu I, Unk I, Haracska L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol 2010; 30:684-93; PMID:19948885; http://dx.doi.org/ 10.1128/MCB.00863-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saugar I, Parker JL, Zhao S, Ulrich HD. The genome maintenance factor Mgs1 is targeted to sites of replication stress by ubiquitylated PCNA. Nucleic Acids Res 2011; 40:245-57; PMID:21911365; http://dx.doi.org/ 10.1093/nar/gkr738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hishida T, Ohno T, Iwasaki H, Shinagawa H. Saccharomyces cerevisiae MGS1 is essential in strains deficient in the RAD6-dependent DNA damage tolerance pathway. EMBO J 2002; 21:2019-29; PMID:11953321; http://dx.doi.org/ 10.1093/emboj/21.8.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, et al. . Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 2012; 47:396-409; PMID:22704558; http://dx.doi.org/ 10.1016/j.molcel.2012.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weston R, Peeters H, Ahel D. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev 2012; 26:1558-72; PMID:22759634; http://dx.doi.org/ 10.1101/gad.193516.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yuan J, Ghosal G, Chen J. The HARP-like domain-containing protein AH2ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol Cell 2012; 47:410-21; PMID:22705370; http://dx.doi.org/ 10.1016/j.molcel.2012.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson RE, Henderson ST, Petes TD, Prakash S, Bankmann M, Prakash L. Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol Cell Biol 1992; 12:3807-18; PMID:1324406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang H, Lawrence CW. The error-free component of the RAD6RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci USA 2005; 102:15954-9; PMID:16247017; http://dx.doi.org/ 10.1073/pnas.0504586102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Branzei D, Vanoli F, Foiani M. SUMOylation regulates Rad18-mediated template switch. Nature 2008; 456:915-20; PMID:19092928; http://dx.doi.org/ 10.1038/nature07587 [DOI] [PubMed] [Google Scholar]
- 43. Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 2005; 19:339-50; PMID:15687257; http://dx.doi.org/ 10.1101/gad.322605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Minca EC, Kowalski D. Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol Cell 2010; 38:649-61; PMID:20541998; http://dx.doi.org/ 10.1016/j.molcel.2010.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Burkovics P, Sebesta M, Balogh D, Haracska L, Krejci L. Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res 2014; 42:1711-20; PMID:24198246; http://dx.doi.org/ 10.1093/nar/gkt1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet 2010; 6:e1001205; PMID:21085632; http://dx.doi.org/ 10.1371/journal.pgen.1001205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, Jentsch S. Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol Cell 2013; 49:536-46; PMID:23260657; http://dx.doi.org/ 10.1016/j.molcel.2012.11.016 [DOI] [PubMed] [Google Scholar]
- 48. Matos J, West SC. Holliday junction resolution: regulation in space and time. DNA Repair 2014; 19:176-81; PMID:24767945; http://dx.doi.org/ 10.1016/j.dnarep.2014.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Prado F, Aguilera A. Control of cross-over by single-strand DNA resection. Trends Genet 2003; 19:428-31; PMID:12902160; http://dx.doi.org/ 10.1016/S0168-9525(03)00173-2 [DOI] [PubMed] [Google Scholar]
- 50. Mankouri HW, Ashton TM, Hickson ID. Holliday junction-containing DNA structures persist in cells lacking Sgs1 or Top3 following exposure to DNA damage. Proc Natl Acad Sci USA 2011; 108:4944-9; PMID:21383164; http://dx.doi.org/ 10.1073/pnas.1014240108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kannouche P, Broughton BC, Volker M, Hanaoka F, Mullenders LH, Lehmann AR. Domain structure, localization, and function of DNA polymerase eta, defective in xeroderma pigmentosum variant cells. Genes Dev 2001; 15:158-72; PMID:11157773; http://dx.doi.org/ 10.1101/gad.187501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, West SC. Human DNA polymerase η promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell 2005; 20:783-92; PMID:16337601; http://dx.doi.org/ 10.1016/j.molcel.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 53. Kawamoto T, Araki K, Sonoda E, Yamashita YM, Harada K, Kikuchi K, Masutani C, Hanaoka F, Nozaki K, Hashimoto N, et al. . Dual roles for DNA polymerase η in homologous DNA recombination and translesion DNA synthesis. Mol Cell 2005; 20:793-9; PMID:16337602; http://dx.doi.org/ 10.1016/j.molcel.2005.10.016 [DOI] [PubMed] [Google Scholar]
- 54. Buisson R, Niraj J, Pauty J, Maity R, Zhao W, Coulombe Y, Sung P, Masson J-Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase h in recombination-associated DNA synthesis at blocked replication forks. Cell Rep 2014; 6:553-64; PMID:24485656; http://dx.doi.org/ 10.1016/j.celrep.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 2010; 17:1305-11; PMID:20935632; http://dx.doi.org/ 10.1038/nsmb.1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010; 141:255-67; PMID:20403322; http://dx.doi.org/ 10.1016/j.cell.2010.02.028 [DOI] [PubMed] [Google Scholar]
- 57. Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010; 465:951-5; PMID:20453836; http://dx.doi.org/ 10.1038/nature09097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. González-Prieto R, Muñoz-Cabello AM, Cabello-Lobato MJ, Prado F. Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J 2013; 32:1307-21; http://dx.doi.org/ 10.1038/emboj.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pagès V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 2003; 300:1300-3; http://dx.doi.org/ 10.1126/science.1083964 [DOI] [PubMed] [Google Scholar]
- 60. Higuchi K, Katayama T, Iwai S, Hidaka M, Horiuchi T, Maki H. Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells 2003; 8:437-49; PMID:12694533; http://dx.doi.org/ 10.1046/j.1365-2443.2003.00646.x [DOI] [PubMed] [Google Scholar]
- 61. McInerney P, O’Donnell M. Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J Biol Chem 2004; 279:21543-51; PMID:15014081; http://dx.doi.org/ 10.1074/jbc.M401649200 [DOI] [PubMed] [Google Scholar]
- 62. García-Gómez S, Reyes A, Martínez-Jiménez MI, Chocrón ES, Mourón S, Terrados G, Powell C, Salido E, Méndez J, Holt IJ, et al. . PrimPol, an archaic primasepolymerase operating in human cells. Mol Cell 2013; 52:541-53; http://dx.doi.org/ 10.1016/j.molcel.2013.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mourón S, Rodriguez-Acebes S, Martínez-Jiménez MI, García-Gómez S, Chocrón S, Blanco L, Méndez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 2013; 20:1383-9; PMID:24240614; http://dx.doi.org/ 10.1038/nsmb.2719 [DOI] [PubMed] [Google Scholar]
- 64. Callegari AJ, Clark E, Pneuman A, Kelly TJ. Postreplication gaps at UV lesions are signals for checkpoint activation. Proc Natl Acad Sci USA 2010; 107:8219-24; PMID:20404181; http://dx.doi.org/ 10.1073/pnas.1003449107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Quinet A, Vessoni AT, Rocha CRR, Gottifredi V, Biard D, Sarasin A, Menck CFM, Stary A. Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA Repair 2014; 14:27-38; PMID:24380689; http://dx.doi.org/ 10.1016/j.dnarep.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 66. Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 2008; 30:519-29; PMID:18498753; http://dx.doi.org/ 10.1016/j.molcel.2008.03.024 [DOI] [PubMed] [Google Scholar]
- 67. Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 1998; 17:598-608; PMID:9430650; http://dx.doi.org/ 10.1093/emboj/17.2.598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Daboussi F, Courbet S, Benhamou S, Kannouche P, Zdzienicka MZ, Debatisse M, Lopez BS. A homologous recombination defect affects replication-fork progression in mammalian cells. J Cell Sci 2008; 121:162-6; PMID:18089650; http://dx.doi.org/ 10.1242/jcs.010330 [DOI] [PubMed] [Google Scholar]
- 69. Henry-Mowatt J, Jackson D, Masson J-Y, Johnson PA, Clements PM, Benson FE, Thompson LH, Takeda S, West SC, Caldecott KW. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol Cell 2003; 11:1109-17; PMID:12718895; http://dx.doi.org/ 10.1016/S1097-2765(03)00132-1 [DOI] [PubMed] [Google Scholar]
- 70. Vázquez MV, Rojas V, Tercero JA. Multiple pathways cooperate to facilitate DNA replication fork progression through alkylated DNA. DNA Repair 2008; 7:1693-704; http://dx.doi.org/ 10.1016/j.dnarep.2008.06.014 [DOI] [PubMed] [Google Scholar]
- 71. Alabert C, Bianco JN, Pasero P. Differential regulation of homologous recombination at DNA breaks and replication forks by the Mrc1 branch of the S-phase checkpoint. EMBO J 2009; 28:1131-41; PMID:19322196; http://dx.doi.org/ 10.1038/emboj.2009.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492-502; PMID:20188668; http://dx.doi.org/ 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yoon J-H, Prakash S, Prakash L. Requirement of Rad18 protein for replication through DNA lesions in mouse and human cells. Proc Natl Acad Sci USA 2012; 109:7799-804; PMID:22547805; http://dx.doi.org/ 10.1073/pnas.1204105109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol 1976; 101:417-25; PMID:1255724; http://dx.doi.org/ 10.1016/0022-2836(76)90156-X [DOI] [PubMed] [Google Scholar]
- 75. Ball LG, Xu X, Blackwell S, Hanna MD, Lambrecht AD, Xiao W. The Rad5 helicase activity is dispensable for error-free DNA post-replication repair. DNA Repair 2014; 16:74-83; PMID:24674630; http://dx.doi.org/ 10.1016/j.dnarep.2014.02.016 [DOI] [PubMed] [Google Scholar]
- 76. Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 2010; 11:683-7; PMID:20842177; http://dx.doi.org/ 10.1038/nrm2974 [DOI] [PubMed] [Google Scholar]
- 77. Yoon D, Wang Y, Stapleford K, Wiesmüller L, Chen J. p53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol 2004; 336:639-54; PMID:15095978; http://dx.doi.org/ 10.1016/j.jmb.2003.12.050 [DOI] [PubMed] [Google Scholar]
- 78. Jossen R, Bermejo R. The DNA damage checkpoint response to replication stress: a game of forks. Front Genet 2013; 4:26; PMID:23493417; http://dx.doi.org/ 10.3389/fgene.2013.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lomonosov M. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 2003; 17:3017-22; PMID:14681210; http://dx.doi.org/ 10.1101/gad.279003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612; http://dx.doi.org/ 10.1016/j.cell.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J 2005; 24:405-17; PMID:15616582; http://dx.doi.org/ 10.1038/sj.emboj.7600511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Feng Z, Zhang J. A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic Acids Res 2012; 40:726-38; PMID:21954437; http://dx.doi.org/ 10.1093/nar/gkr748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tian F, Sharma S, Zou J, Lin S-Y, Wang B, Rezvani K, Wang H, Parvin JD, Ludwig T, Canman CE, et al. . BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc Natl Acad Sci USA 2013; 110:13558-63; PMID:23901102; http://dx.doi.org/ 10.1073/pnas.1306534110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gonzalez-Huici V, Szakal B, Urulangodi M, Psakhye I, Castellucci F, Menolfi D, Rajakumara E, Fumasoni M, Bermejo R, Jentsch S, et al. . DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J 2014; 33:327-40; PMID:24473148; http://dx.doi.org/ 10.1002/embj.201387425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ulrich HD. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair 2009; 8:461-9; PMID:19217833; http://dx.doi.org/ 10.1016/j.dnarep.2009.01.006 [DOI] [PubMed] [Google Scholar]
- 86. Parnas O, Zipin-Roitman A, Pfander B, Liefshitz B, Mazor Y, Ben-Aroya S, Jentsch S, Kupiec M. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J 2010; 29:2611-22; PMID:20571511; http://dx.doi.org/ 10.1038/emboj.2010.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kubota T, Nishimura K, Kanemaki MT, Donaldson AD. Short article. Mol Cell 2013; 50:273-80; PMID:23499004; http://dx.doi.org/ 10.1016/j.molcel.2013.02.012 [DOI] [PubMed] [Google Scholar]
- 88. Lee KY, Yang K, Cohn MA, Sikdar N, D’Andrea AD, Myung K. Human ELG1 Regulates the Level of Ubiquitinated Proliferating Cell Nuclear Antigen (PCNA) through Its Interactions with PCNA and USP1. J Biol Chem 2010; 285:10362-9; PMID:20147293; http://dx.doi.org/ 10.1074/jbc.M109.092544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Falbo KB, Alabert C, Katou Y, Wu S, Han J, Wehr T, Xiao J, He X, Zhang Z, Shi Y, et al. . Involvement of a chromatin remodeling complex in damage tolerance during DNA replication. Nat Struct Mol Biol 2009; 16:1167-72; PMID:19855395; http://dx.doi.org/ 10.1038/nsmb.1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)M phase rather than S phase. Proc Natl Acad Sci USA 2006; 103:8971-6; PMID:16751278; http://dx.doi.org/ 10.1073/pnas.0510167103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lin J-R, Zeman MK, Chen J-Y, Yee M-C, Cimprich KA. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell 2011; 42:237-49; PMID:21396873; http://dx.doi.org/ 10.1016/j.molcel.2011.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gangavarapu V, Haracska L, Unk I, Johnson RE, Prakash S, Prakash L. Mms2-Ubc13-dependent and -independent roles of Rad5 ubiquitin ligase in postreplication repair and translesion DNA synthesis in saccharomyces cerevisiae. Mol Cell Biol 2006; 26:7783-90; PMID:16908531; http://dx.doi.org/ 10.1128/MCB.01260-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gangavarapu V, Santa Maria SR, Prakash S, Prakash L. Requirement of replication checkpoint protein kinases Mec1Rad53 for postreplication repair in yeast. mBio 2011; 2:e00079-11; PMID:21586645; http://dx.doi.org/ 10.1128/mBio.00079-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pages V, Santa Maria SR, Prakash L, Prakash S. Role of DNA damage-induced replication checkpoint in promoting lesion bypass by translesion synthesis in yeast. Genes Dev 2009; 23:1438-49; PMID:19528320; http://dx.doi.org/ 10.1101/gad.1793409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Flott S, Kwon Y, Pigli YZ, Rice PA, Sung P, Jackson SP. Regulation of Rad51 function by phosphorylation. EMBO Rep 2011; 12:833-9; PMID:21738226; http://dx.doi.org/ 10.1038/embor.2011.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Conde F, Ontoso D, Acosta I, Gallego-Sánchez A, Bueno A, San-Segundo PA. Regulation of tolerance to DNA alkylating damage by Dot1 and Rad53 in Saccharomyces cerevisiae. DNA Repair 2010; 9:1038-49; PMID:20674515; http://dx.doi.org/ 10.1016/j.dnarep.2010.07.003 [DOI] [PubMed] [Google Scholar]
- 97. Naiman K, Philippin G, Fuchs RP, Pages V. Chronology in lesion tolerance gives priority to genetic variability. Proc Natl Acad Sci USA 2014; 111:5526-31; PMID:24706928; http://dx.doi.org/ 10.1073/pnas.1321008111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer 2001; 1:22-33; PMID:11900249; http://dx.doi.org/ 10.1038/35094000 [DOI] [PubMed] [Google Scholar]
- 99. Novarina D, Amara F, Lazzaro F, Plevani P, Muzi-Falconi M. Mind the gap: keeping UV lesions in check. DNA Repair 2011; 10:751-9; PMID:21602108; http://dx.doi.org/ 10.1016/j.dnarep.2011.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352-5; PMID:18323444; http://dx.doi.org/ 10.1126/science.1140735 [DOI] [PubMed] [Google Scholar]
- 101. Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999; 399:700-4; PMID:10385124; http://dx.doi.org/ 10.1038/21447 [DOI] [PubMed] [Google Scholar]
- 102. Avkin S, Sevilya Z, Toube L, Geacintov N, Chaney SG, Oren M, Livneh Z. p53 and p21 regulate error-prone DNA repair to yield a lower mutation load. Mol Cell 2006; 22:407-13; PMID:16678112; http://dx.doi.org/ 10.1016/j.molcel.2006.03.022 [DOI] [PubMed] [Google Scholar]
- 103. Moinova HR, Chen W-D, Shen L, Smiraglia D, Olechnowicz J, Ravi L, Kasturi L, Myeroff L, Plass C, Parsons R, et al. . HLTF gene silencing in human colon cancer. Proc Natl Acad Sci USA 2002; 99:4562-7; PMID:11904375; http://dx.doi.org/ 10.1073/pnas.062459899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sandhu S, Wu X, Nabi Z, Rastegar M, Kung S, Mai S, Ding H. Loss of HLTF function promotes intestinal carcinogenesis. Mol Cancer 2012; 11:18; PMID:22452792; http://dx.doi.org/ 10.1186/1476-4598-11-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Motegi A, Sood R, Moinova H, Markowitz SD, Liu PP, Myung K. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J Cell Biol 2006; 175:703-8; PMID:17130289; http://dx.doi.org/ 10.1083/jcb.200606145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Myung K, Smith S. The RAD5-dependent postreplication repair pathway is important to suppress gross chromosomal rearrangements. JNCI Monographs 2008; 2008:12-5; PMID:18647995; http://dx.doi.org/ 10.1093/jncimonographs/lgn019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Bell DW, Sikdar N, Lee K-Y, Price JC, Chatterjee R, Park H-D, Fox J, Ishiai M, Rudd ML, Pollock LM, et al. . Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLoS Genet 2011; 7:e1002245; PMID:21901109; http://dx.doi.org/ 10.1371/journal.pgen.1002245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bishop A. Role of homologous recombination in carcinogenesis. Exp Mol Pathol 2003; 74:94-105; PMID:12710940; http://dx.doi.org/ 10.1016/S0014-4800(03)00010-8 [DOI] [PubMed] [Google Scholar]
- 109. Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 2010; 329:82-5; PMID:20595613; http://dx.doi.org/ 10.1126/science.1191125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A. Break-induced replication is highly inaccurate. Plos Biol 2011; 9:e1000594; PMID:21347245; http://dx.doi.org/ 10.1371/journal.pbio.1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. . ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 2013; 27:1610-23; PMID:23873943; http://dx.doi.org/ 10.1101/gad.214080.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 2003; 426:870-4; PMID:14685245; http://dx.doi.org/ 10.1038/nature02253 [DOI] [PubMed] [Google Scholar]
- 113. Janz C, Wiesmüller L. Wild-type p53 inhibits replication-associated homologous recombination. Oncogene 2002; 21:5929-33; PMID:12185593; http://dx.doi.org/ 10.1038/sj.onc.1205757 [DOI] [PubMed] [Google Scholar]
- 114. Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response. Cell 2004; 118:699-713; PMID:15369670; http://dx.doi.org/ 10.1016/j.cell.2004.08.015 [DOI] [PubMed] [Google Scholar]
- 115. Barlow JH, Rothstein R. Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase. EMBO J 2009; 28:1121-30; PMID:19262568; http://dx.doi.org/ 10.1038/emboj.2009.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Meister P, Taddei A, Vernis L, Poidevin M, Gasser SM, Baldacci G. Temporal separation of replication and recombination requires the intra-S checkpoint. J Cell Biol 2005; 168:537-44; PMID:15716375; http://dx.doi.org/ 10.1083/jcb.200410006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Prado F. Genetic instability is prevented by Mrc1-dependent spatio-temporal separation of replicative and repair activities of homologous recombination. Bioessays 2014; 36:451-62; PMID:24615940; http://dx.doi.org/ 10.1002/bies.201300161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mankouri HW, Ngo H-P, Hickson ID. Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1-Rmi1-Top3. Mol Biol Cell 2007; 18:4062-73; PMID:17671161; http://dx.doi.org/ 10.1091/mbc.E07-05-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sollier J, Driscoll R, Castellucci F, Foiani M, Jackson SP, Branzei D. The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol Biol Cell 2009; 20:1671-82; PMID:19158389; http://dx.doi.org/ 10.1091/mbc.E08-08-0875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Branzei D, Sollier J, Liberi G, Zhao X, Maeda D, Seki M, Enomoto T, Ohta K, Foiani M. Ubc9- and Mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 2006; 127:509-22; PMID:17081974; http://dx.doi.org/ 10.1016/j.cell.2006.08.050 [DOI] [PubMed] [Google Scholar]