

Abstract
Antiangiogenic agents targeting the vascular endothelial growth factor A (VEGFA) pathway play an important role in current cancer treatment modalities but are limited by alternative angiogenesis mechanisms. Recent studies suggest that enhanced signaling through a COX-2/PGE2 axis contributes to VEGF-independent tumor angiogenesis. Thus, COX-2/PGE2 inhibition may potentiate VEGF therapies.
Keywords: angiogenesis, axitinib, bevacizumab, celecoxib, cyclooxygenase-2 (COX-2), endothelial, metastasis, non-steroidal anti-inflammatory drugs (NSAIDs), prostaglandin E2 (PGE2), vascular endothelial growth factor (VEGF)
Abbreviations
- AA
arachidonic acid
- COX
cyclooxygenase
- COXIB
selective COX-2 inhibitor
- HPLC
high-performance liquid chromatography
- NSAID
non-steroidal anti-inflammatory drug
- PG
prostaglandin
- PGDH
15-prostaglandin dehydrogenase
- PTGES
prostaglandin E synthase
- RTK
receptor tyrosine kinase
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
Angiogenesis is a critical hallmark of malignant tumor growth. Analogous to normal tissue development, blood vessels provide expanding tumors with vital oxygen and nutrients and generate an outlet for the removal of metabolic waste products. The newly formed blood vessels also provide an escape route for metastasis. Vascular endothelial growth factor A (VEGFA), the most widely recognized stimulator of angiogenesis, is an important driver of cancer and other angiogenesis-dependent diseases such as macular degeneration. Several agents that block the VEGF pathway have been approved by the US Food and Drug Administration, including bevacizumab, an anti-VEGF neutralizing antibody; aflibercept, a soluble decoy receptor or “VEGF trap”; and seven small molecule receptor tyrosine kinase (RTK) inhibitors, namely axitinib, cabozantinib, pazopanib, regorafenib, sorafenib, sunitinib, and vandetanib. Among the RTK inhibitors, axitinib is the most selective for VEGFR1, VEGFR2, and VEGFR3 at clinically used doses. Antiangiogenic agents generally work best in combination with chemotherapy and have been found to extend the lives of patients with colon, lung, or kidney cancer. Unfortunately, however, improvements in survival are typically modest, measuring a few months at best. Even while on VEGF blocking therapy the tumors can continue to grow and angiogenesis persists through ill-defined VEGF-independent mechanisms.1 Persistent tumor angiogenesis is also observed in animal tumor models, stimulating many laboratories, including ours, to search for alternative angiogenesis pathways that are potentially responsible for clinical refractoriness to VEGF therapies.
To identify alternative angiogenesis mechanisms we focused on CT26 colon tumors, which at the outset of our studies appeared to evoke angiogenesis predominantly through the secretion of a putative angiogenesis factor that was VEGF-independent.2 A bioactive factor detectable in the supernatant of cultured CT26 cells was purified to homogeneity from 10 liters of conditioned medium using multiple cycles of reverse phase HPLC and was identified by mass spectrometry as prostaglandin E2 (PGE2). PGE2 is derived from arachidonic acid through a series of enzymatic reactions, of which cyclooxygenase 2 [prostaglandin endoperoxide synthase 2 (PTGS2), best known as COX-2] is generally rate limiting (Fig. 1). After 2 weeks of treatment with celecoxib, a COX-2 selective inhibitor, CT26 tumor volume in mice was blocked by 93%. To further investigate the role of COX-2 we used a variant of the human HCT-116 colon cancer cell line, HCT-VKO, in which both alleles of the VEGF gene were disrupted by homologous recombination. HCT-VKO cells, which expressed low endogenous levels of COX-2 protein, were poorly tumorigenic upon subcutaneous injection into immunodeficient mice. However, tumor angiogenesis and tumor growth could be rescued by exogenous overexpression of either Vegf or COX-2. Although overexpression of either gene promoted tumor growth, careful evaluation of the respective pathways revealed independent underlying mechanisms. For example, phosphorylation of Vegfr2 in tumors was blocked by treatment with axitinib but was unaffected by celecoxib. COX-2 expression, on the other hand, was inhibited by celecoxib but not axitinib. Furthermore, VEGF blockers were most effective against VEGF-driven tumors whereas celecoxib was most effective against COX-2 driven tumors.2
Figure 1.
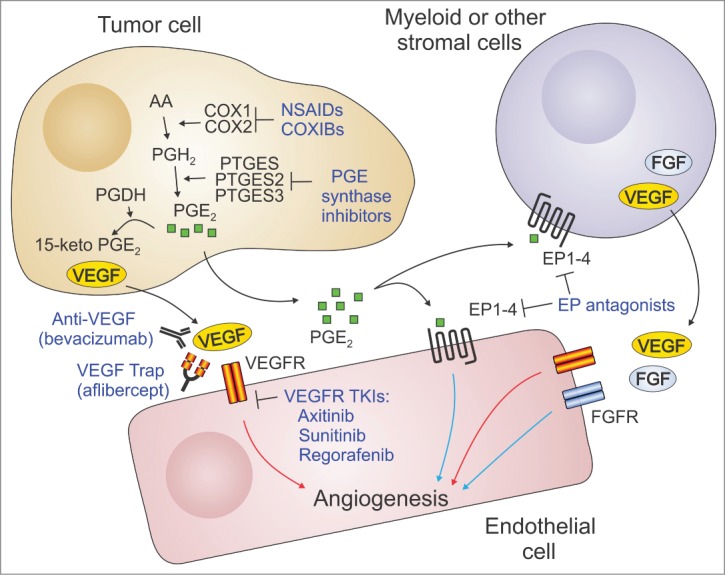
VEGF-dependent and -independent mechanisms of endothelial cell activation. Vascular endothelial growth factor A (VEGFA) produced by tumor cells can directly stimulate angiogenesis through the activation of VEGF receptors (VEGFRs) on endothelial cells. PGE2 produced by tumor cells can also activate angiogenesis through direct stimulation of the G-protein coupled receptors EP1 to EP4 on endothelial cells. PGE2 can also promote angiogenesis indirectly by recruiting proangiogenic myeloid cells and other inflammatory cells or fibroblasts to the tumors. These cells produce various angiogenic stimulators such as fibroblast growth factor (FGF) and VEGF. VEGF signaling can be blocked by anti-VEGF neutralizing antibodies (bevacizumab), soluble VEGF receptors (aflipercept), or several VEGFR tyrosine kinase inhibitors (TKIs). PGE2 signaling can be blocked through the use of NSAIDs, COXIBs, PGE synthase inhibitors, or EP receptor antagonists. Because of the independence of the pathways, the most effective inhibition of angiogenesis may result from simultaneous targeting of both the COX-2/PGE2 and VEGF pathways. Red arrows: VEGF-dependent angiogenesis; Blue arrows: VEGF-independent angiogenesis. AA: arachidonic acid; COX: cyclooxygenase; PG: prostaglandin; PGDH: 15-prostaglandin dehydrogenase; PTGES: prostaglandin E synthase.
Based on the independent mechanisms of these pathways in promoting tumor angiogenesis and tumor growth, we hypothesized that simultaneous targeting of the COX-2 and VEGF pathways may improve antiangiogenic activity. Indeed, compared to the respective monotherapies, dual pathway inhibition reduced angiogenesis and growth of colon (CT26 or HCT116) or breast (4T1) tumors. More importantly, dual COX-2/VEGF pathway blockade was much more effective than the monotherapies at blocking experimental HCT-116 colon cancer liver metastasis and spontaneous 4T1 breast cancer metastasis. In the 4T1 breast cancer model, initiation of therapy after surgical resection of the primary orthotopic tumors blocked the outgrowth of metastases and significantly enhanced overall survival, suggesting that the combination therapy may be useful in the adjuvant setting.2
Non-steroidal anti-inflammatory drugs (NSAIDs) that target both COX-1 and COX-2 are widely used to treat pain, fever, and inflammation and are also well known in the oncology field as potential chemopreventive agents. In most tissues, COX-2 is an inducible enzyme whose expression is elevated during inflammation and cancer whereas COX-1 is constitutively expressed. COX-1 inhibition is thought to be responsible for the rare but serious gastrointestinal and renal toxicities that can occur with long-term use of NSAIDs. COXIBs that are selective for COX-2 were initially developed to overcome such toxicities, but were later found to increase the risk of cardiovascular events. Importantly, celecoxib may be less cardiotoxic than rofecoxib (Vioxx) and safe for use in patients with a low baseline cardiovascular risk.3 In patients at higher risk, aspirin or naproxen may be a suitable alternative in combination with proton pump inhibitors to help prevent gastrotoxicities.4 PGE2 synthase inhibitors or PGE2 receptor antagonists also represent promising drug alternatives that may help circumvent the toxicities associated with COX inhibitors (Fig. 1). However, these agents are currently at an early stage of clinical development.5-6
Antiangiogenic agents were clinically approved for cancer treatment based on their ability to prolong overall survival in patients with advanced metastatic disease. These successes stimulated hundreds of clinical trials to determine whether VEGF blockers could also help prevent recurrence and metastasis.7 So far, however, these trials have failed to demonstrate any benefit of adding VEGF blockers in the perioperative adjuvant or neoadjuvant setting.8 For some cancers, activation of the COX-2/PGE2 pathway may help to explain this refractoriness to VEGF therapy; however, other VEGF-independent angiogenic mechanisms may also be involved.1 Therefore, to maximize efficacy and minimize toxicities it will be important to identify which tumors utilize both VEGF- and COX-2/PGE2-dependent pathways. In the case of the COX-2/PGE2 pathway, examining tumor biopsies for overexpression of COX-2 protein may help, although inactivating mutations in the 15-hydroxyprostaglandin dehydrogenase (PGDH) gene, which encodes the enzyme responsible for PGE2 catabolism, can also lead to elevated PGE2 levels.9
PGDH-inactivating mutations may also lead to resistance to COX-2 selective inhibitors, perhaps because constitutive levels of COX-1 and/or residual activity of COX2 are sufficient in this context to increase local PGE2 levels.9 Activating mutations in PIK3CA, on the other hand, may lead to elevated expression of COX-2.10 Thus, tumors that maintain wild type PGDH and harbor activating PIK3CA mutations and high COX-2 levels may be particularly sensitive to COX-2 inhibition. Although progress in identifying VEGF-responsive tumors has proven even more challenging, our studies with CT26 tumors suggest that blocking alternative angiogenesis pathways such as the COX-2/PGE2 pathway has the potential to unmask VEGF inhibitor sensitivity that may not be detectable otherwise.2
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Supported by the Center for Cancer Research Intramural Program, NCI, NIH, part of the US Department of Health and Human Services (DHHS). The content of this publication does not necessarily reflect the views or policies of the DHHS.
References
- 1. Abdollahi A, Folkman J. Evading tumor evasion: current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist Updat 2010; 13:16-28; PMID:20061178; http://dx.doi.org/ 10.1016/j.drup.2009.12.001 [DOI] [PubMed] [Google Scholar]
- 2. Xu L, Stevens J, Hilton MB, Seaman S, Conrads TP, Veenstra TD, Logsdon D, Morris H, Swing DA, Patel NL, et al. . COX-2 Inhibition Potentiates Antiangiogenic Cancer Therapy and Prevents Metastasis in Preclinical Models. Sci Transl Med 2014; 6:242ra84; PMID:24964992; http://dx.doi.org/ 10.1126/scitranslmed.3008455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang D, DuBois RN. The role of anti-inflammatory drugs in colorectal cancer. Annu Rev Med 2013; 64:131-44; PMID:23020877; http://dx.doi.org/ 10.1146/annurev-med-112211-154330 [DOI] [PubMed] [Google Scholar]
- 4. Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, Egger M, Jüni P. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ 2011; 342:c7086; PMID:21224324; http://dx.doi.org/ 10.1136/bmj.c7086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther 2013; 138:485-502; PMID:23523686; http://dx.doi.org/ 10.1016/j.pharmthera.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang HH, Meuillet EJ. Identification and development of mPGES-1 inhibitors: where we are at? Future Med Chem 2011; 3:1909-34; PMID:22023034; http://dx.doi.org/ 10.4155/fmc.11.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011; 8:210-21; PMID:21364524; http://dx.doi.org/ 10.1038/nrclinonc.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oyan B. Why do targeted agents not work in the adjuvant setting in colon cancer? Expert Rev Anticancer Ther 2012; 12:1337-45; PMID:23176621; http://dx.doi.org/ 10.1586/era.12.111 [DOI] [PubMed] [Google Scholar]
- 9. Yan M, Myung SJ, Fink SP, Lawrence E, Lutterbaugh J, Yang P, Zhou X, Liu D, Rerko RM, Willis J, et al. . 15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc Natl Acad Sci U S A 2009; 106:9409-13; PMID:19470469; http://dx.doi.org/ 10.1073/pnas.0902367106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, et al. . Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 2012; 367:1596-606; PMID:23094721; http://dx.doi.org/ 10.1056/NEJMoa1207756 [DOI] [PMC free article] [PubMed] [Google Scholar]