

Abstract
Increased O-GlcNAcylation is emerging as a general characteristic of cancer cells that is critical for multiple oncogenic phenotypes. Recently, we demonstrated that elevated O-GlcNAcylation contributes to the metabolic shift seen in cancer through stabilization of the glycolytic regulator HIF-1α and links metabolism to stress and cancer cell survival.
Keywords: cancer, epithelial, ER stress, GLUT1, hexosamine biosynthetic pathway, HIF-1α, metabolism, O-GlcNAcylation, OGT
Tumor cells display a metabolic shift termed the “Warburg Effect” that increases glucose uptake and glycolytic flux. These metabolic changes play an essential role in supporting oncogenic phenotypes by regulating cancer cell growth and survival.1 Cancer cells exploit this mechanism to produce energy through high rates of glucose uptake and glycolysis, thus allowing maximum production of amino acids, lipids, and nucleotides to fuel the increase in biomass that is essential for cancer cell proliferation. Oncogenes and tumor suppressors drive the shift to aerobic glycolysis seen in cancer cells in part through activation of the mammalian target of rapamycin (mTOR) pathway, which promotes translation of transcription factors known to directly regulate nutrient uptake and metabolism, including hypoxia-inducible factor 1a (HIF-1α).1,2 These changes in glucose uptake by tumor cells increase flux into glycolysis as well as divergent pathways including the hexosamine biosynthetic pathway (HBP). Approximately 2–5% of glucose enters the HBP and, along with glutamine, generates the activated sugar nucleotide uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) that can serve as a substrate for the enzyme O-GlcNAc transferase (OGT). This results in the covalent addition of O-linked GlcNAc moieties onto serine and threonine residues of a wide range of nuclear and cytosolic proteins and exhibits analogous functions to phosphorylation, influencing protein–protein interactions and protein stability and function.3 Thus, nutrient-sensitive O-GlcNAcylation modulates transcriptional events and cellular signaling pathways in mammalian cells.3
Recently, our group was the first to show that breast and prostate cancer cells contain elevated levels of OGT and O-GlcNAcylation compared to their normal counterparts.4,5 Normalization of OGT and O-GlcNAc levels in cancer cells through RNAi or pharmacological inhibition induces cell cycle arrest, reduces proliferation, and decreases primary tumor growth in vivo. Furthermore, inhibition of OGT reduces breast and prostate cancer cell invasion and metastasis both in vitro and in vivo.5 Since alterations in O-GlcNAc levels have also been associated with metabolic disorders such as diabetes, we hypothesized that O-GlcNAcylation might play a role in the altered metabolism seen in cancer cells. Our recent study demonstrated that OGT and O-GlcNAcylation promote the Warburg effect and survival in cancer cells through regulation of HIF-1α stability.6 Using metabolomics analysis, we found that reducing OGT levels using RNAi resulted in an overall decrease in the production of lactate, as well as other carbohydrate intermediates. Consistent with reversal of the Warburg effect, targeting OGT resulted in an overall increase in tricarboxylic acid (TCA) cycle intermediates including α-ketoglutarate (α-KG), indicating that OGT/O-GlcNAcylation regulates the Warburg effect in breast cancer cells.
HIF-1α is known to be a major regulator of cancer cell metabolism by directly controlling the gene expression of glycolytic enzymes.2 Breast cancer cells with reduced OGT levels or activity contained decreased HIF-1α protein levels and decreased expression of its transcriptional gene targets including glucose transporter 1 (GLUT1), lactate dehydrogenase A (LDHA), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), and phosphofructokinase-1 (PFK-1). OGT regulation of HIF-1α protein was proteasome-dependent, since OGT inhibition increased HIF-1α ubiquitination, increased the interaction between HIF-1α and its E3 ligase pVHL, and increased HIF-1α hydroxylation. Thus, OGT regulates HIF-1α protein degradation in a proteasome- and pVHL-dependent manner to promote the Warburg effect in cancer cells. Results from liquid chromatog-raphy–mass spectrometry (LC-MS) showed that α-KG levels were significantly increased in response to targeting OGT. We further showed that OGT-mediated regulation of HIF-1α hydroxylation and stability occurs, in part, through modulation of α-KG, a required co-factor for this hydroxylation event. We observed that restoring HIF1α levels under conditions of OGT depletion by introduction of a hydroxylation-resistant non-degradable HIF1α mutant reverses the metabolic defects induced by OGT reduction in breast cancer cells. These data suggest that OGT regulation of α-KG levels contributes to modulation of HIF-1α hydroxylation to promote cancer cell glycolytic flux (Fig. 1).
Figure 1.
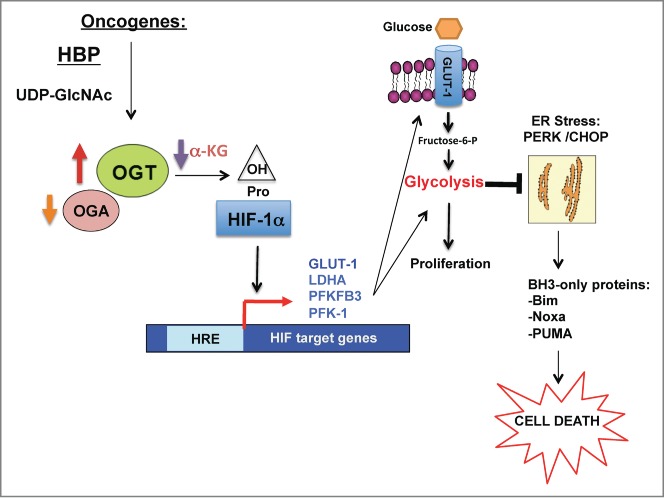
OGT links cancer cell metabolism and cell survival via regulation of HIF-1α. Schematic depicting the pathway by which oncogenes increase OGT (and decrease OGA) levels and decrease production of α-KG, a key metabolite that regulates HIF-1α hydroxylation and stabilization. Once HIF-1α is stabilized, it increases transcription of key glycolytic enzymes including GLUT1 that increase glycolytic flux, and blocks ER stress-mediated signals and apoptotic factors including Bcl-2 family proteins such as Bim, thus contributing to cancer cell survival. α-KG, α-ketoglutarate; Bim, Bcl2-interacting mediator of cell death; CHOP, C/EBP homologous protein; Fructose-6-P, fructose-6-phosphate; GLUT1, glucose transporter 1; HBP, hexosamine biosynthetic pathway; HIF-1α, hypoxia inducible factor 1α; HRE, HIF-1 response element; LDHA, lactate dehydrogenase A; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; OH, hydroxyl group; PERK, PRKR-like endoplasmic reticulum kinase; PFK-1, phosphofructokinase-1; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; Pro, proline; PUMA, p53-upregulated modulator of apoptosis; UDP-GlcNAc, uridine diphosphate N-acetylglucosamine.
Cancer cells have been shown to be sensitive to metabolic stress and undergo cell death upon antiglycolytic treatment. Specifically, decreased cellular energy has been associated with activation of the unfolded protein response (UPR) in various cancer types. Consistent with a decrease in glucose uptake and increased metabolic stress, we observed a selective induction of endoplasmic reticulum (ER) stress-induced cell death in breast cancer cells upon OGT inhibition. Additionally, we observed that this bioenergetic stress and cell death effect was dependent on the regulation of HIF-1α hydroxylation by OGT. We also investigated whether overexpression of GLUT1, a HIF-1α transcriptional target, was sufficient to rescue the glycolytic and survival defects induced by targeting OGT. Indeed, GLUT1 overexpression rescued glycolytic and survival defects and partially rescued tumor growth in vivo. Collectively, these data indicate that regulation of the HIF-1α signaling axis by OGT contributes to the glycolytic shift and associated cell survival seen in cancer (Fig. 1).
The HBP and O-GlcNAcylation have recently garnered much attention regarding the regulation of cell survival and tumorigenesis. Several reports have demonstrated that targeting various enzymes in the HBP, including OGT, can inhibit survival of oncogene-driven and hypoxic cancer cells. Specifically, targeting the rate-limiting enzyme glutamine fructose 6-phosphate amidotransferase (GFPT1) results in reduction of pancreatic tumor growth in a Kras-driven model.7 In regard to metabolic regulation, Itkonen et al. recently demonstrated that OGT can stabilize another major metabolic transcription factor, c-MYC, to promote prostate cancer cell growth.8 Our work shows that O-GlcNAcylatoin is a key regulator not only of glycolytic metabolites, but also of metabolites in the TCA cycle including a-KG. Since overexpression of GLUT1 was able to reverse the effects of OGT knockdown on glycolysis, as well as a-KG levels,6 we hypothesize that regulation of the HIF1/GLUT1 axis by O-GlcNAcylation is central to cancer cell metabolism. However, it is also possible that OGT directly or indirectly regulates mitochondrial proteins to fine tune mitochondrial metabolism associated with the TCA cycle. Recent studies have shown that many mitochondrial proteins are O-GlcNAcylated and that modification of specific mitochondrial proteins may affect mitochondrial cellular responses.9 Identification of novel O-GlcNAc-regulated metabolites and specific targets will advance our understanding of the role of O-GlcNAcylation in metabolism in both normal and cancer cells. Consistent with our results showing that O-GlcNAcylation regulates ER-stress induced cell death in cancer cells, very recent reports have shown a close relationship between HBP/O-GlcNAcylation and ER stress associated with aging and myocardial infarction.10 Thus O-GlcNAcylation is positioned as a key sensor that links metabolism, cell stress, and cell survival pathways. Further elucidation of how O-GlcNAcylation intersects with cellular stress responses will help identify novel strategies to combat disease states.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Valerie Sodi for critically reading this commentary.
Funding
This work is supported by NCI grants CA183574 (to CMF) and CA155413 (to MJR).
References
- 1. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11-20; PMID:18177721; http://dx.doi.org/ 10.1016/j.cmet.2007.10.002 [DOI] [PubMed] [Google Scholar]
- 2. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 2013; 123:3664-71; PMID:23999440; http://dx.doi.org/ 10.1172/JCI67230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 2011; 80:825-58; PMID:21391816; http://dx.doi.org/ 10.1146/annurev-biochem-060608-102511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, Vosseller K, Reginato MJ. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 2010; 29:2831-42; PMID:20190804; http://dx.doi.org/ 10.1038/onc.2010.41 [DOI] [PubMed] [Google Scholar]
- 5. Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem; 2012. 287:11070-81; PMID:22275356; http://dx.doi.org/ 10.1074/jbc.M111.302547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, Vocadlo DJ, Seagroves TN, Reginato MJ. O-GlcNAcylation Regulates Cancer Metabolism and Survival Stress Signaling via Regulation of the HIF-1 Pathway. Mol Cell 2014; 54:820-31; PMID:24857547; http://dx.doi.org/ 10.1016/j.molcel.2014.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. . Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149:656-70; PMID:22541435; http://dx.doi.org/ 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, Svindland A, Schlomm T, Mills IG. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res 2013; 73:5277-87; PMID:23720054; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0549 [DOI] [PubMed] [Google Scholar]
- 9. Palaniappan KK, Hangauer MJ, Smith TJ, Smart BP, Pitcher AA, Cheng EH, Bertozzi CR, Boyce M. A chemical glycoproteomics platform reveals O-GlcNAcylation of mitochondrial voltage-dependent anion channel 2. Cell Rep 2013; 5:546-52; PMID:24120863; http://dx.doi.org/ 10.1016/j.celrep.2013.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vincenz L, Hartl FU. Sugarcoating ER Stress. Cell 2014; 156:1125-7; PMID:24630714; http://dx.doi.org/ 10.1016/j.cell.2014.02.035 [DOI] [PubMed] [Google Scholar]