

Abstract
Chemoresistance remains a major impediment in cancer therapy. Although major progress has been made in understanding the mechanisms underlying resistance in cancer, there is still more to learn. Our studies provide evidence that Gli1 drives a novel form of drug resistance involving Phase II drug metabolism enzymes, specifically the UGT1A family.
Keywords: cancer, drug resistance, Gli1, UGT1A, glucuronidation, Vismodegib, cytarabine, ribavirin, eIF4E
If you heard the many stories featuring the struggle between cancer treatment, resistance, and relapse throughout the history of cancer research, you might assume that we are very familiar with the topic. Our new data suggest that this is not the case.
While studying the efficacy of ribavirin, a drug that targets eIF4E activity, in acute myeloid leukemia (AML) patients with poor prognosis, we found ourselves dealing with what appears to be the inevitable outcome of targeted monotherapy treatments: clinical relapse and molecular resistance.1 During our attempt to understand the molecular events underlying drug resistance in our patients, we serendipitously identified a novel form of drug resistance with implications well beyond ribavirin.2 This new form of resistance involves the well-known Sonic Hedgehog transcription factor Gli1 and the drug modifying enzymes UDP-glucuronysltransferase 1A (UGT1A), and represents a new method by which cancer cells combine the effects of genetic rewiring and metabolic inactivation to evade therapy and allow relapse (Fig. 1). Our story depicts Gli1 alone as a sufficient driver of the glucuronidation of several drugs used for the treatment of AML, including cytarabine, the cornerstone of treatment, and ribavirin. Glucuronidation is a process by which drugs are chemically modified by the addition of glucuronic acid, which typically enhances their hydrophilicity and increases efflux.3 However, in some cases, such as with androgens, glucuronidation alters targeting, rather than drug efflux.4 In this case, glucuronidation of ribavirin correlates with loss of its interaction with eIF4E and ultimately leads to the loss of growth inhibition with no effect on drug uptake.
Figure 1.
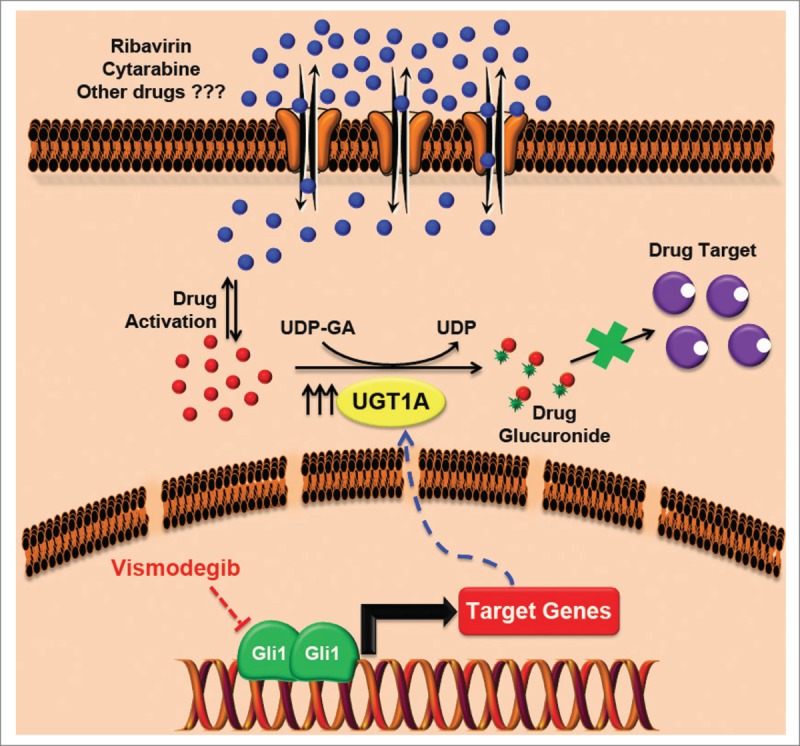
Novel mechanism of multidrug resistance driven by Gli1. Elevated Gli1 levels increase the levels of UGT1As, which in turn catalyze the addition of glucuronic acid to the drug thus leading to the loss of drug–target interaction. This mechanism of resistance applies to ribavirin, cytarabine, and possibly some other drugs and can be reverted by direct or upstream inhibitors of Gli1 such as vismodegib. Note that both ribavirin and cytarabine require an activation step in order to remain in the cell. Our in vitro studies suggest that, at least for ribavirin, glucuronidation occurs on the active form. Further studies are required to assess whether the activation step a priori is essential for glucuronidation to occur or whether this is specific to the drug and/or the UGT1A protein involved.
Fortunately, this new mechanism is clinically promising for patients with AML, for whom the overall survival is on average 7 months. Using vismodegib, a FDA-approved inhibitor of the Sonic Hedgehog pathway, we showed that targeting Gli1 eliminated ribavirin or cytarabine glucuronidation, allowed the re-emergence of eIF4E-ribavirin complexes, and led to renewed drug sensitivity. Importantly, Gli1 inhibition alone had little effect on cell growth whereas Gli1 inhibition in combination with either ribavirin or cytarabine reverted the drug resistance and restored sensitivity. To date, Gli1 inhibitors have been used for basal cell carcinomas in which the Gli1 pathway is inappropriately activated.5 Here, we propose a new rationale for targeting the Gli1 pathway to overcome Gli1-inducible drug glucuronidation. A clinical trial combining vismodegib with ribavirin for the treatment of AML is scheduled to open in the near future (ClinicalTrials.gov NCT02073838). Our findings thus far leave us wondering whether this mechanism could also mediate therapeutic resistance in other types of cancer and which drug families might be targets of Gli1-mediated glucuronidation.
Very little is known about the factors regulating the production of UGT1A enzymes.6 Our studies to date suggest that Gli1 controls the protein stability of UGT1A rather than its transcription. It is also likely that UGT1A expression and activity are regulated by mechanisms other than Gli1-mediated control, a possibility that must be assessed. Furthermore, our studies suggest that ribavirin and cytarabine are glucuronidated by different members of the UGT1A family. Thus, determining the specific member(s) driving resistance is essential to guide the development of selective and specific UGT1A inhibitors in future avenues of research.
To date, the major focus in drug resistance has been on (1) impaired uptake or increased drug efflux as characterized by the multidrug resistant (MDR) drug transporters,7,8 (2) impaired pro-drug metabolism,8 (3) mutation of target proteins e.g., BCR-Abl and Gleevec or PML-RARA and retinoic acid,9 and (4) altered cellular pathways, such as downregulation of target proteins, enhanced DNA repair, activation of alternative survival pathways, inactivation of apoptotic pathways, or activation of antiapoptotic pathways.10 This new mechanism of Gli1-mediated resistance and relapse proves that we are still at the very beginning of understanding and overcoming cancer resistance.
In summary, drug resistance is the underlying reason for treatment failure in the majority of cancer patients. Moreover, development of resistance to one therapy often suggests that patients will be resistant to other therapies that they have never been exposed to. Typically, strategies to treat cancer are based on identifying an Achilles’ heel in the cancer cell, and when resistance emerges the treatment choices become limited and typically do not account for why the resistance emerged in the first place. Perhaps a better approach to the development of more effective and durable cancer therapies would be to determine the underlying causes of treatment failure and success.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Assouline S, Culjkovic B, Cocolakis E, Rousseau C, Beslu N, Amri A, Caplan S, Leber B, Roy DC, Miller WH, Jr, et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof-of-principle clinical trial with ribavirin. Blood 2009; 114:257-60; PMID:19433856; http://dx.doi.org/ 10.1182/blood-2009-02-205153 [DOI] [PubMed] [Google Scholar]
- 2. Zahreddine HA, Culjkovic-Kraljacic B, Assouline S, Gendron P, Romeo AA, Morris SJ, Cormack G, Jaquith JB, Cerchietti L, Cocolakis E, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014; 511:90-3; PMID:24870236; http://dx.doi.org/ 10.1038/nature13283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cummings J, Ethell BT, Jardine L, Boyd G, Macpherson JS, Burchell B, Smyth JF, Jodrell DI. Glucuronidation as a mechanism of intrinsic drug resistance in human colon cancer: reversal of resistance by food additives. Cancer Res 2003; 63:8443-50; PMID:14679008 [PubMed] [Google Scholar]
- 4. Chouinard S, Yueh MF, Tukey RH, Giton F, Fiet J, Pelletier G, Barbier O, Bélanger A. Inactivation by UDP-glucuronosyltransferase enzymes: the end of androgen signaling. J Steroid Biochem Mol Biol 2008; 109:247-53; PMID:18467088; http://dx.doi.org/ 10.1016/j.jsbmb.2008.03.016 [DOI] [PubMed] [Google Scholar]
- 5. LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res 2011; 17:2502-11; PMID:21300762; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther 2005; 106:97-132; PMID:15781124; http://dx.doi.org/ 10.1016/j.pharmthera.2004.10.013 [DOI] [PubMed] [Google Scholar]
- 7. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006; 5:219-34; PMID:16518375; http://dx.doi.org/ 10.1038/nrd1984 [DOI] [PubMed] [Google Scholar]
- 8. Jordheim LP, Dumontet C. Review of recent studies on resistance to cytotoxic deoxynucleoside analogues. Biochim Biophys Acta 2007; 1776:138-59; PMID:17881132 [DOI] [PubMed] [Google Scholar]
- 9. Lovly CM, Shaw AT. Molecular pathways: resistance to kinase inhibitors and implications for therapeutic strategies. Clin Cancer Res 2014; 20:2249-56; PMID:24789032; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol 2013; 4:28; PMID:23504227 [DOI] [PMC free article] [PubMed] [Google Scholar]