

Abstract
There is currently immense interest in understanding the biological consequences of aberrant necroptosis. The recently uncovered role for X-linked inhibitor of apoptosis protein (XIAP) in blocking tumor necrosis factor-dependent necroptosis explains, at least in part, the systemic hyperinflammatory syndrome XLP-2. However, it also points to rather unexpected differences between XIAP and the related proteins baculoviral IAP repeat containing 2 and 3 (cIAP1/2).
Keywords: inflammation, necroptosis, RIP3, XIAP, cell death, XLP-2, cIAP1, cIAP2
Our study
It has become increasingly clear that the manner of cell death and the resulting inflammatory responses are tightly linked processes.1 Cell death by programmed necrosis (necroptosis) elicits potent inflammatory responses, whereas apoptosis is considered largely immunologically silent.
Our recent observation that X-linked inhibitor of apoptosis protein (XIAP, also known as BIRC4) mediates nuclear factor kappa-B (NF-κB) activation in the nucleotide-binding oligomerization domain containing 2 (NOD2) signaling pathway suggested a proinflammatory function of XIAP in this context.2 In striking contrast, XIAP-deficient patients who are diagnosed with the primary immune deficiency syndrome X-linked lymphoproliferative disease-2 (XLP-2) often respond to common viral infections with systemic hyperinflammation, which points to an anti-inflammatory function of XIAP.3 Recent data suggest that this condition is substantially underdiagnosed due to the lack of a simple diagnostic test.4 We hypothesized that NOD2-independent inflammatory processes must be affected when XIAP is absent or non-functional.
Not surprisingly, we found that loss of XIAP led to increased death of innate immune cells such as macrophages and dendritic cells when these cells were activated by tumor necrosis factor (TNF). However, this cell death turned out to be caspase-independent. In contrast, TNF-driven cell death of Xiap−/− dendritic cells and macrophages was completely blocked by genetic deletion of receptor-interacting serine-threonine kinase 3 (RIPK3). Dendritic cells from a mutant knock-in mouse line expressing only a RING-deleted form of XIAP (XIAPΔRING), which was generated by Hermann Steller's lab, clearly demonstrated that XIAP was acting as an E3-ligase.5
The capability to modulate TNF-dependent cell death and act independently of caspase inhibition pointed toward a molecular functionality of XIAP similar to that of baculoviral IAP repeat containing 2 and 3 (cIAP1/2). However, in contrast to cIAP1/2, XIAP had never been observed within the TNF receptor complex I. This suggested distinct functions of XIAP and cIAP1/2, which was emphasized by the fact that depletion of cIAP1/2 in WT dendritic cells using an IAP antagonist did not reproduce the same phenotype as that observed in XIAP-deficient cells.5 In line with this, we found that XIAP controlled the coordinated ubiquitylation of RIPK within a secondary TNF-dependent protein complex containing RIPK3.
The elevated level of necroptosis in XIAP-deficient cells explains some aspects of the XLP-2 syndrome; however, a critical contribution toward explaining the hyperinflammation was the observation that Xiap−/− cells readily activated the NLR family, pyrin domain containing 3 (Nlrp3) inflammasome when treated with TNF or Toll-like receptor (TLR) ligands. This TNF-dependent process resulted in the secretion of large amounts of the proinflammatory cytokine IL-1β and a systemic hyperinflammatory phenotype in mice, similar to the human hyperinflammatory syndrome XLP-2.3,5 The assembly of the inflammasome in Xiap−/− cells was again downstream of RIPK3, implicating RIPK3 activation as a central signaling node for initiating inflammation.
Open Questions
Our study raised several unanswered questions. In the absence of XIAP, we observed increased catalytic activity of caspase-8 upon TNF treatment. Curiously, the enhanced processing of caspase-8 was blocked when RIP3 was co-deleted, which indicated that caspase-8 processing was downstream of RIPK3 activation, or at least within the same complex. A recent study has shown that deletion of caspase-8 in dendritic cells (using the CD11c-Cre system) led to increased IL-1β secretion in response to TLR4.6 However, the mechanism reported appears to be distinct from the phenotype that we observe in Xiap−/− bone marrow-derived dendritic cells (BMDCs), as our caspase-8−/− DCs did not die upon TLR4 activation.5 The exact function of caspase-8 in our system therefore remains elusive (Fig. 1).
Figure 1.
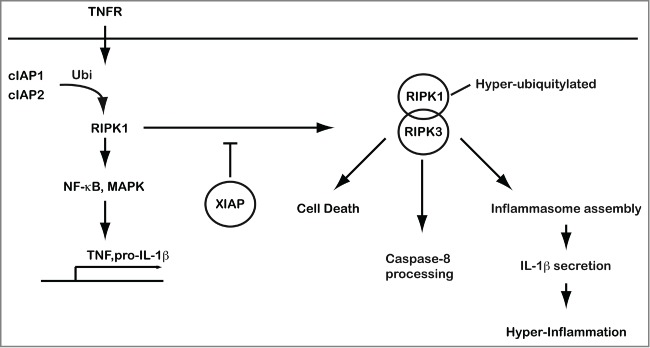
XIAP-mediated suppression of RIPK3-dependent cell death and inflammation. XIAP prevents activation of RIPK3 that drives cell death and triggers inflammasome assembly and activation. XIAP controls the ubiquitylation of RIPK1 in a complex with RIPK3 in response to activation of the TNFR1 complex. RIPK, receptor-interacting serine-threonine kinase 3; TNFR, tumor necrosis factor receptor; XIAP, X-linked inhibitor of apoptosis protein.
In addition, our work points to a strictly RIPK3-dependent assembly of the inflammasome in XIAP-deficient cells. Direct association of RIPK3 with the inflammasome has not been reported and the mechanism of inflammasome activation in response to RIP3 activation or necroptosis remains unanswered. One possible mechanism is indirect activation mediated by the action of mixed lineage kinase domain-like protein (MLKL) at the plasma membrane.7 Activated MLKL induced by RIPK3-dependent phosphorylation could form pores that allow K+ influx, which subsequently triggers assembly of the Nlrp3 inflammasome. Interestingly, a recent report shows that Ripk3−/−caspase-8−/− macrophages are unable to produce pro–IL-1β in response to TLR4 activation, which points toward a priming defect.8 Together, these data indicate that RIPK3 and caspase-8 are important for priming and activation of the inflammasome, but further work is required to fully understand the underlying molecular mechanisms.
Implications
IAP antagonists used in clinical trials for both hematological and solid cancers were initially designed to broadly inhibit XIAP and cIAP1/2.9 Because of the dramatic effect that these molecules have on the stability of cIAP1/2, particular attention has since been focused on the role of these 2 proteins in mediating TNF-dependent cell death. Although maximal induction of cell death in response to IAP antagonists requires inhibition of all 3 IAPs, XIAP has been thought to act primarily as a bona fide caspase inhibitor.9 Our study, however, indicates that XIAP plays a significant role as an ubiquitin ligase in suppressing TNF-dependent cell death in innate immune cells. The antitumor efficacy of IAP antagonists might therefore involve the modulation of immune responses, as exemplified by the fact that IAP antagonists have also been shown to augment T-cell responses.10
Another implication of our work is the critical role of TNF in triggering disease pathology in XLP-2 patients. Currently, the XLP-2 syndrome is rarely diagnosed, but recent studies indicate that XIAP mutations are also found in (mostly pediatric) patients diagnosed with early-onset inflammatory bowel disease such as Crohn's and other undefined pathologies.4 Our work supports the notion that patients with XIAP mutations might profit from anti-TNF therapies such as infliximab or etanercept when used early during an inflammatory episode.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Murphy JM, Silke J. Ars Moriendi; the art of dying well - new insights into the molecular pathways of necroptotic cell death. EMBO Reports 2014; 15:155-64; PMID:24469330; http://dx.doi.org/ 10.1002/embr.201337970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, Rieser E, Rickard JA, Bankovacki A, Peschel C, et al. . The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mole Cell 2012; 46:746-58; PMID:22607974; http://dx.doi.org/ 10.1016/j.molcel.2012.04.014 [DOI] [PubMed] [Google Scholar]
- 3. Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, Bleesing JJ, Zhang K, Filipovich AH. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood 2010; 116:1079-82; PMID:20489057; http://dx.doi.org/ 10.1182/blood-2010-01-256099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Speckmann C, Lehmberg K, Albert MH, Damgaard RB, Fritsch M, Gyrd-Hansen M, Rensing-Ehl A, Vraetz T, Grimbacher B, Salzer U, et al. . X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol 2013; 149:133-41; PMID:23973892; http://dx.doi.org/ 10.1016/j.clim.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 5. Yabal M, Muller N, Adler H, Knies N, Gross CJ, Damgaard RB, Kanegane H, Ringelhan M, Kaufmann T, Heikenwalder M, et al. . XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep 2014; 7:1796-808; PMID:24882010; http://dx.doi.org/ 10.1016/j.celrep.2014.05.008 [DOI] [PubMed] [Google Scholar]
- 6. Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013; 38:27-40; PMID:23260196; http://dx.doi.org/ 10.1016/j.immuni.2012.09.015 [DOI] [PubMed] [Google Scholar]
- 7. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol Cell. 2014; 54(1):133-46; http://dx.doi.org/ 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 8. Allam R, Lawlor KE, Yu EC, Mildenhall AL, Moujalled DM, Lewis RS, Ke F, Mason KD, White MJ, Stacey KJ, et al. . Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep 2014; 15(9):982-90 PMID:24990442; http://dx.doi.org/ 10.15252/embr.201438463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Dis 2012; 11:109-24; PMID:22293567; http://dx.doi.org/ 10.1038/nrd3627 [DOI] [PubMed] [Google Scholar]
- 10. Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, Li W, Neuberg D, Blumberg R, et al. . IAP inhibitors enhance co-stimulation to promote tumor immunity. J Exp Med 2010; 207:2195-206; PMID:20837698; http://dx.doi.org/ 10.1084/jem.20101123 [DOI] [PMC free article] [PubMed] [Google Scholar]