

Abstract
DNA double-strand breaks (DSBs) are highly lethal lesions that jeopardize genome integrity. However, DSBs are also used to generate diversity during the physiological processes of meiosis or establishment of the immune repertoire. Therefore, DSB repair must be tightly controlled. Two main strategies are used to repair DSBs: homologous recombination (HR) and non-homologous end joining (NHEJ). HR is generally considered to be error-free, whereas NHEJ is considered to be error-prone. However, recent data challenge these assertions. Here, we present the molecular mechanisms involved in HR and NHEJ and the recently described alternative end-joining mechanism, which is exclusively mutagenic. Whereas NHEJ is not intrinsically error-prone but adaptable, HR has the intrinsic ability to modify the DNA sequence. Importantly, in both cases the initial structure of the DNA impacts the outcome. Finally, the consequences and applications of these repair mechanisms are discussed. Both HR and NHEJ are double-edged swords, essential for maintenance of genome stability and diversity but also able to generate genome instability.
Keywords: DNA repair, double-strand break repair, genome instability, genome rearrangements, homologous recombination, ionizing radiation, mutagenesis, non-homologous end joining, telomeres
Abbreviations
- A-EJ
alternative end joining
- BIR
break-induced replication
- BRIM
break repair-induced mutation
- CDK
cyclin-dependent kinase
- CG
gene conversion
- CO
crossing over
- CSR
class switch recombination
- DDR
DNA damage response
- DSB
double-strand break
- DSBR
double-strand break repair
- FoSTeS
fork stalling and template switching
- HR
homologous recombination
- INDEL
insertion/deletion
- IR
ionizing radiation
- MMBIR
microhomology-mediated BIR
- MRN
MRE11/RAD50/NBS1
- NAHR
non-allelic homologous recombination
- NHEJ
non-homologous end joining
- C-NHEJ
canonical non-homologous end joining
- PNK
polynucleotide kinase
- SCE
sister chromatid exchange
- SDSA
synthesis-dependent strand annealing
- SNP
single nucleotide polymorphism
- SSA
single-strand annealing
- TdT
terminal deoxynucleotidyl transferase
Introduction
DNA double-strand breaks (DSBs) are very harmful lesions that can trigger profound genome rearrangements leading to cell death, senescence, developmental abnormalities, and both tumor initiation and progression. Although maintenance of genome stability is vital, genetic diversity is essential in certain physiological processes such as generation of the immune repertoire or the recombination of parental alleles during meiosis. The absence of genetic diversity leads to an evolutionary dead end. In these cases, programmed DSBs are physiologically produced by controlled cellular enzymes. Furthermore, telomeres are specific structures at the ends of the chromosomes that can be erroneously recognized as DSBs.
Therefore, DNA repair must maintain genomic stability, while allowing genetic diversity. Strikingly, the most prominent DSB repair mechanisms exhibit a duality that allows them to both maintain and alter genome stability.
The DSB Repair Processes
DSBs can be repaired by two main strategies: homologous recombination (HR) or non-homologous end joining (NHEJ). Homologous recombination requires a homologous DNA sequence and actually represents different processes involving both common and distinct mechanisms (Fig. 1). NHEJ involves ligation of the DNA ends of a DSB without requiring any homologous sequence (Fig. 2). Notably, additional alternative end-joining pathway(s) have recently been described.
Figure 1.
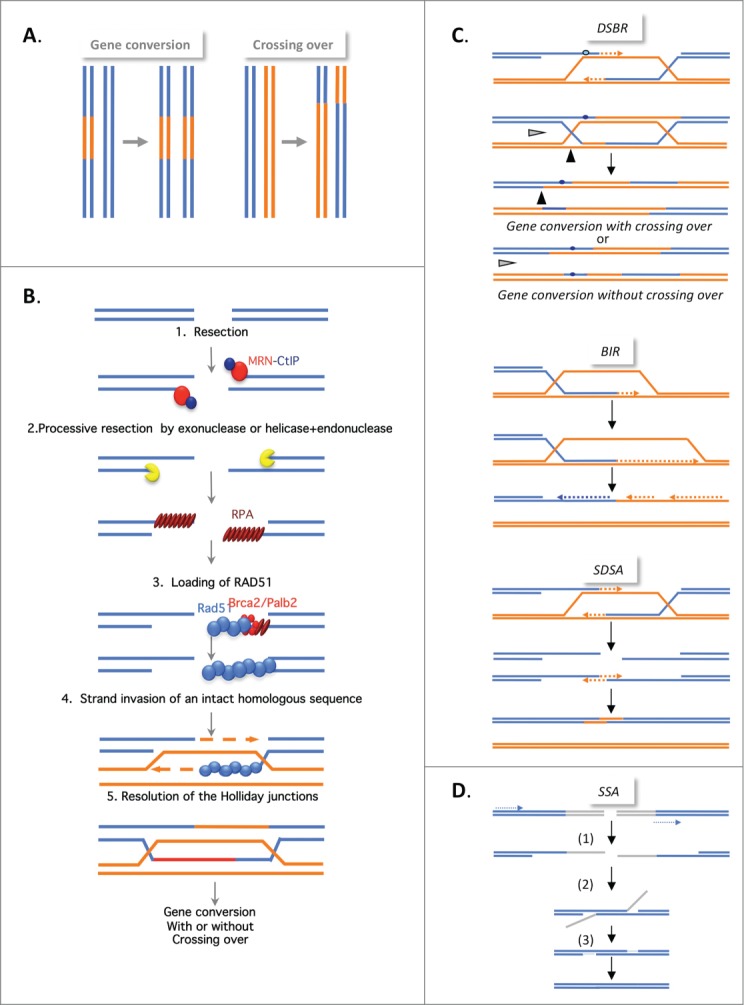
(See previous page). Mechanisms of homologous recombination. (A) The products of HR. Gene conversion (GC; left panel), results from the non-reciprocal exchange of a DNA sequence (in orange). Crossing over (CO; right panel), results from the reciprocal exchange of DNA sequences (orange and blue). Both GC and CO are outcomes of the HR events described below. (B) The double strand break repair model (DSBR). HR is initiated by 5′ to 3′ single-stranded resection of double-stranded DNA ends through the action of different protein complexes, MRN/CtIP, Exo1, and BLM/TopoIII/RMI, acting in 2 steps. BRCA1, in association with CtIP, favors resection initiation through the removal of 53BP1. This resection creates a 3′ single-stranded DNA (ssDNA) that is coated with replication protein A (RPA). A complex including Palb2 and BRCA2, which are both breast tumor suppressors, then replaces RPA with Rad51. The Rad51 nucleoprotein filament invades the intact homologous duplex DNA, priming DNA synthesis, and the intact DNA molecule1 is copied, creating a D-Loop (displacement loop). This process tolerates limited polymorphisms, thus creating heteroduplex intermediates bearing mismatches (blue circle in corresponding left panel in Fig. 1C). This step generates 2 cruciform intermediates also known as Holliday junctions. (C) Different HR mechanisms. In the DSBR model (upper panel), the heteroduplex molecules are represented by the blue circles. Strand invasion and DNA synthesis can lead to GCs. Depending on how the Holliday junctions are resolved, the GC will be associated with CO (black arrow) or not (gray arrow). In synthesis-dependent strand annealing (SDSA, middle panel), the initiation is similar but the invading strand dehybridizes and reanneals at the other end of the broken molecule and no Holliday junction is generated. In break-induced replication (BIR, lower panel), DNA synthesis occurs over longer distances, even reaching the end of the chromosome. Here, there is neither resolution of the intermediates nor crossing over. (C) Single-strand annealing (SSA). An extended single-strand resection (1) reveals 2 complementary ssDNA strands; hybridization of the 2 complementary strands (2) generates an intermediate; resolution (3) and gap filling complete the repair. This process can occur between 2 homologous sequences in tandem in the same orientation (dotted arrows) and results in deletion of the intergenic sequences.
Figure 2.

(See previous page). Mechanisms of non-homologous end joining. (A). Canonical NHEJ (C-NHEJ). C-NHEJ is initiated by binding of the Ku80-Ku70 heterodimer to the DSB, which recruits the DNA-PK catalytic subunit, DNA-PKcs. Several proteins, including Artemis, the polynucleotide kinase (PNK) and members of the polymerase X family, process the DNA ends to make them competent for the subsequent ligation steps. Finally, ligase IV, in association with Xrcc4 and Cernunos/Xlf, seals the double-strand ends. (B) Alternative end-joining (A-EJ). In the absence of Ku70/Ku80, the DNA ends are resected in a reaction favored by the nuclease activity of Mre11 and CtIP. Note that Parp1 is involved in A-EJ initiation. The resulting ssDNA reveals complementary microhomologies (2–4 nt or more) that can be annealed; gap filling completes the end joining. Finally, Xrcc1 and ligase III complete the A-EJ process. Notably, A-EJ is always associated with deletions at the junctions and frequently, but not systematically, involves microhomologies that are distant from the DSB. (C) The 2-step DSB repair pathway choice model. After signaling of the DSB by ATM and MRN, (I) binding of Ku80/Ku70 protects the DSB ends from resection, routing DSB repair toward the conservative C-NHEJ pathway. (II) The nuclease activity of Mre11 and CtIP favor ssDNA resection, which can then initiate the HR or A-EJ pathway. A short ssDNA resection is sufficient for A-EJ but not for HR. A-EJ is an exclusively non-conservative mutagenic process.
Homologous recombination
The products of HR are gene conversions (GCs, non-reciprocal exchange of genetic material) associated with or without crossing over (CO, reciprocal exchange of the adjacent sequences) (Fig. 1A). All of the different HR processes are initiated by 5′ to 3′ single-stranded resection of the double-stranded DNA ends, creating a 3′ single-stranded DNA (ssDNA) end. Strand invasion promoted by RAD51 (Fig. 1B) generates cruciform intermediates called Holliday junctions. At this point, the HR mechanisms differ in the processing of the intermediates, leading to gene conversion associated with or without crossing over, synthesis-dependent strand annealing (SDSA), or break-induced replication (BIR) (Figs. 1B and C).1
Another process, single-strand annealing (SSA), is also initiated by resection; however, the subsequent steps do not involve strand invasion of an intact DNA duplex (and are therefore RAD51-independent) but instead involve the annealing of 2 complementary ssDNA sequences (Fig. 1D). Note that SSA is exclusively a non-conservative process.
Another crucial role of HR in the maintenance of genome stability is escorting replication fork progression; HR efficiently seals ssDNA gaps when replication forks reach DNA lesions and is also involved in the recovery of arrested replication forks.2
Non-homologous end joining and alternative end joining pathways
The first end joining pathway identified, canonical-NHEJ (C-NHEJ), is initiated by the binding of Ku80–Ku70 heterodimer to the DSB, which then recruits the DNA-PK catalytic subunit, DNA-PKcs. Finally, ligase IV, in association with its cofactors (XRCC4 and XLF/Cernunos), seals the double-strand ends (Fig. 2A).1
Recently, the existence of at least one alternative end-joining pathway has emerged (Fig. 2B). Different names have been used in the literature for this alternative pathway: alternative end joining (A-EJ or alt-NHEJ), backup NHEJ (B-NHEJ), and microhomology-mediated end joining (MMEJ). To distinguish this alternative pathway from C-NHEJ, and because some repair events do not use microhomologies, we will refer to this alternative pathway as A-EJ (for review see3). The characteristics of the A-EJ pathway are as follows: (1) It does not require extended homologous sequences; (2) Unlike C-NHEJ, it is independent of Ku80 and Xrcc4/Ligase IV; (3) It is associated with deletions at the repair junction, frequently (but not systematically) using microhomologies distant from the DSB. Notably, 2 different classes of microhomologies that are used for re-sealing DSBs should be distinguished: microhomologies at the DSB itself are used by C-NHEJ, whereas microhomologies distant from the DSB are used by A-EJ.3
The signature of A-EJ led to the model shown in Fig. 2B in which A-EJ is initiated by ssDNA resection. The resection step shares some characteristics with HR, such as the involvement of MRE11 nuclease activity and CtIP, which are counteracted by 53BP1 and RIF1 (for a review see3). However, the resection length required for A-EJ is much shorter (a few nucleotides are sufficient) than that required for HR. These characteristics suggest a 2-step model for the choice between the different DSB repair pathways3,4 (Fig. 2C).
Resection at DNA ends: impact on the choice of DSB repair pathway
Protection of DNA ends
A key step in choosing the appropriate repair pathway is the decision between protection versus resection of DNA ends (Fig. 2C). Several proteins have been implicated in protection, but the main factor involved is the Ku70/Ku80 heterodimer (for review see3). Consistent with this role, defects in Ku80/Ku70 result in robust stimulation of HR.5
One role of BRCA1-CtIP in HR initiation is to displace 53BP1 from the DNA ends, thereby enabling ssDNA resection.6 Notably, CtIP can also displace Ku from DNA ends.7
Physiologically, 53BP1 deficiency leads to profound defects in class switch recombination (CSR) and V(D)J recombination (for review see8). At non-V(D)J or CSR junctions, 53BP1 is directly implicated in DNA end protection during A-EJ.9
In addition, RIF1 plays a key role in DNA end protection in mammals and is epistatic to 53BP1.9-12 Both proteins are also involved in the fusion of uncapped telomeres via NHEJ10,12 and, more generally, protect against long resections in A-EJ.9 The binding of RIF1 to 53BP1 requires phosphorylation of 53BP1 on a subset of ST/Q sites by ataxia telangiectasia mutated (ATM) kinase.
A second 53BP1-interacting protein, PTIP, has been suggested to limit resection. Like RIF1, it binds ST/Q motifs in the N-terminal domain of 53BP1.13,14 Mutation of the PTIP binding sites for 53BP1 or deletion of PTIP does not affect CSR but abrogates illicit NHEJ in BRCA1-deficient cells treated with PARP inhibitors.13
Resection and its regulation by CDKs
The process of end resection can be divided into 2 successive steps.15 A key regulator of resection is the MRN (Mre11, Rad50, Nbs1) complex. MRE11 initiates resection for HR, A-EJ, and CSR.4,16,17 in association with the nuclease CtIP.18 The endonuclease activity of MRE11 initiates resection and the exonuclease activity extends the resection, at least in the case of HR.19
CtIP and MRN are sufficient for short-range resection, but to generate longer ssDNA tracts other nuclease activities are required. Two distinct sets of nucleases and their associated protein components, Exo1 or Dna2 with BLM (also known as Sgs1 in yeast)/Top3/Rmi1, are required for long-range end resection (for review see15). Robust cell cycle regulation is achieved by the cyclin-dependent protein kinases (CDKs). CDK2-dependent phosphorylation of S327 on CtIP is a prerequisite for CtIP/BRCA1 interaction and occurs only in the S/G2 phase.20,21 MRE11 associates with both CKD2 and CtIP and facilitates this phosphorylation.22 In addition to phosphorylation on S327, phosphorylation of T847 on CtIP by CDK is required to promote resection.23
Recently, phosphorylation of Exo1 by CDK1/2 and its impact on resection was also demonstrated.24
Dual-function players
BRCA1 is found in several different complexes (for review see25) that have opposite roles in DNA end resection: MRN/CtIP/BRCA1 favors resection whereas the BRCA1/RAP80/ BRCC36/ABRAXAS complex inhibits resection and promotes C-NHEJ. Moreover, BRCA1 interacts with Ku80 and stabilizes its association with DNA ends.26 Strikingly, both complexes are formed mainly in the S and G2 phases of the cell cycle.
Another dual-function player is the BLM (mutated in Bloom syndrome) protein. BLM has been implicated in resection together with TopoIII/Rmi1 and Dna2.27,28 However, BLM also protects DNA ends against deletions during A-EJ.9 Interestingly, BLM is differentially associated with 53BP1 in G1 and with TopoIII in S/G2, thereby providing a switch between its protective and pro-resection activities.9 This switch favors protection when HR is not proficient and triggers long-range resections in S/G2 when HR should be favored.
Choice of the DSB repair pathway: the 2-step competition model
The choice of an appropriate DSB repair pathway, in particular with respect to the phase of the cell cycle, is crucial for the maintenance of genome stability. This choice is made in 2 steps: (1) C-NHEJ versus resection, and (2) HR versus A-EJ (Fig. 2C). In the first step, Ku represses both HR and A-EJ (for review see3). Several parameters can affect the second step including the presence of a homologous sequence, the size of the resection, and the cell cycle phase. Indeed, HR is only active during the S and G2 phases, whereas both C-NHEJ and A-EJ are active throughout the cell cycle (for review see3,29).
The Accuracy of DSB Repair
A-EJ and SSA are exclusively non-conservative pathways. Furthermore, A-EJ has been proposed to promote chromosome translocation.30,31
In the literature, HR is generally considered to be error-free whereas NHEJ is considered to be error-prone. However, based on more recent data, it is necessary to revise this current view.
HR and genetic stability
Consistent with the involvement of HR in the maintenance of genome stability, cells defective in HR exhibit increased mutagenesis and genome rearrangements (for review see29).
Replication stress impacts not only the accuracy of DNA replication but also chromosome segregation. Indeed, endogenous or low levels of replication stress induce mitotic defects, including anaphase bridges, supernumerary centrosomes, and multipolar mitosis, which lead to uneven chromosome segregation and aneuploidy.32 Because HR plays a pivotal role in the resumption of arrested replication forks, defects in HR lead to spontaneous slowed replication fork progression, anaphase bridges, common fragile sites, and supernumerary centrosomes, which result in multipolar mitosis and aneuploidy (for review see2,29). These data underline the essential role played by HR in protecting genome stability at the interface between replication and mitosis.
HR and genetic instability
Because HR can generate heteroduplex molecules and exchange adjacent sequences through GC and CO, it is intrinsically able to alter genetic information through several mechanisms.
The intrinsic capacity of HR to generate genetic modifications
Strand invasion between 2 DNA molecules with few heterologous sequences generates heteroduplexes bearing mismatches (Fig. 1C). Subsequent mismatch repair can lead to genetic modifications on the recipient molecule. In addition, a sequence that was absent in the invading molecule can be copied during DNA synthesis initiated by the 3′ end of the invading strand, thereby transferring this genetic information to the repaired molecule in a gap-repair-like process (see Figs. 1B and C).
When HR occurs between fully identical sequences (for example, during sister chromatid exchange [SCE]), it does not typically impact the genetic material. However, unequal SCE can lead to sequence duplications or deletions (Fig. 3A). The fact that unequal SCEs are less frequent than equal SCEs contributes to the maintenance of genome stability and therefore HR is frequently considered to be error free. However, one can argue that this is not due to the accuracy of the HR process itself, but simply because the DNA strands are identical (as a result of accurate DNA replication), because HR is restricted to S and G2 phases, which correspond to the cell cycle phases in which sister chromatids are present, and because the tight cohesion of the sister chromatids channels HR toward equal SCE. Thus, the structure of the DNA and associated mechanisms, rather than the HR process itself, favors an error-free outcome. During meiosis, which aims to generate genetic diversity, SCEs are repressed and HR between homologous chromosomes (which are not fully identical) is favored. The necessity to strictly control HR through the associated processes reflects the intrinsic potential danger of HR. In addition, even with fully identical molecules, HR can initiate mutagenic DNA synthesis (see below).
Figure 3.
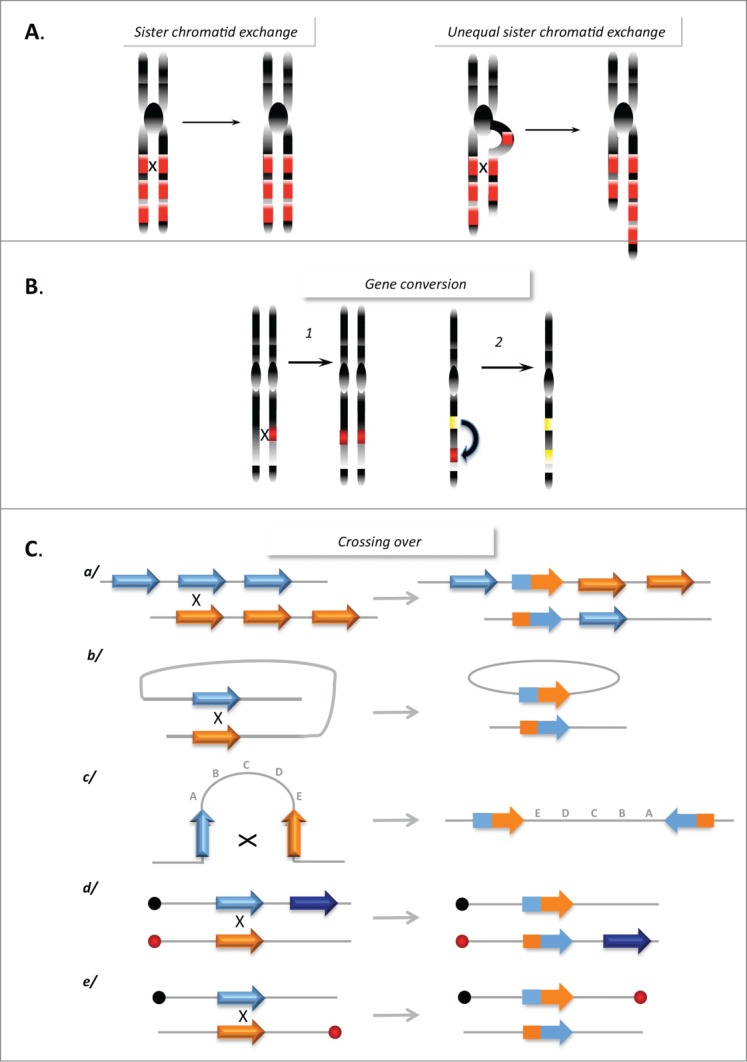
Genome instability promoted by HR. (A) Sister chromatid exchanges. Left panel: Equal SCE between repeat sequences (red boxes) does not affect genome stability. Right panel: Unequal SCEs, leading to amplification and loss of genetic material. (B) Gene conversion between 2 heteroalleles (left panel) leading to a loss of heterozygosity, or between a pseudogene (yellow box) and a gene (red box) leading to inactivation of the gene. (C) Rearrangements resulting from CO between repeat sequences: (a) Between homologous sequences on 2 chromosomes, leading to amplification and deletion; (b) Intramolecular CO between direct repeats, resulting in the excision of the intervening sequence; (c) Intramolecular CO between 2 inverted repeats, resulting in inversion of the internal fragment; (d and e) Interchromosomal CO generates a translocation (d) or a dicentric and an acentric chromosome (e) depending on the position relative to their respective centromeres (black and red circles).
Moreover, GC between 2 heteroalleles can transfer genetic information in a non-reciprocal manner, resulting in loss of heterozygosity. GC between a pseudogene and a related coding sequence can transfer stop codons, resulting in inactivation of the coding gene (Fig. 3B). In addition, genomic rearrangements can be generated through CO between repeated sequences that are dispersed throughout the genome (Fig. 3C) (for review see29).
Considering the events described above, the maintenance of genome stability requires repression of HR in the G1 phase and restriction of HR to equal SCEs during the S and G2 phases.
Genome rearrangements mediated by BIR and MMBIR
Replicative stress induced by the overexpression of cyclin E leads to copy number alterations. Many replicative stress-induced duplications have been attributed to BIR events or to microhomology-mediated replication (MMBIR), a BIR-related mechanism. The authors of these studies proposed that repair of damaged replication forks through BIR accounts for the segmental genomic duplications observed in human cancers and that the larger amplifications (> 200 kb) and deletions arise from other repair mechanisms, such as non-allelic homologous recombination (NAHR).33
Error-prone DNA synthesis initiated by HR
Mutagenesis associated with HR was first reported in E. coli. In budding yeast, mutagenic HR-mediated DSB repair has been referred to as break-repair-induced mutation (BRIM) (reviewed in29). More recently, it has been shown that the mutation rate of DNA synthesis initiated by GC is enhanced up to 1,400-fold.34 Additionally, DNA synthesis induced by BIR is highly inaccurate over the entire path of the replication fork35. Finally, in fission yeast, replication restart by HR not only mediates NAHR but is also associated with error-prone DNA synthesis.36
Protection against HR toxicity
In mitotic cells, the orientation toward SCE avoids potential HR-mediated genetic instability as discussed above. In addition to genome mutagenesis and rearrangements (see above), excessive HR can lead to the accumulation of unresolved HR intermediates, which themselves generate genomic instability and cell death.37 Therefore, the HR process must be tightly controlled to avoid unnecessary HR events.
Helicases destabilize abortive HR intermediates and thus protect against genomic instability.38 Moreover, limitation of unscheduled HR initiation should also prevent the accumulation of toxic HR intermediates. In mammalian cells, this type of protective role in limiting HR initiation has been proposed for p53, Bcl-2, and AKT1.39-41
Importantly, the fact that these protective systems have evolved to prevent excess HR underlines the potential risks of this pathway.
The accuracy of NHEJ
For a long time, NHEJ was considered error prone. However, since the discovery of A-EJ, which is distinct from C-NHEJ and highly error-prone, the apparent infidelity of C-NHEJ must be re-evaluated.3
C-NHEJ and genetic stability
At the chromosomal level, C-NHEJ is essential for genome stability. Indeed, deficiencies in C-NHEJ result in profound genomic rearrangements. C-NHEJ also prevents trinucleotide repeat fragility and expansion.42 Conversely, A-EJ is involved in chromosome translocation.30
The repair of DSBs induced by ionizing radiation (IR) or V(D)J recombination requires processing of the DNA ends prior to ligation. Thus, in these cases DNA end processing, rather than the C-NHEJ process per se, generates the mutagenesis/variability. Therefore, biological systems that do not require DNA end processing are necessary to address the actual accuracy of C-NHEJ.
The DNA ends generated by endonucleases are biochemically competent for direct ligation without requiring a preliminary “cleaning” step. Two situations can occur: (1) the overhang ends are fully complementary, or (2) the overhanging ends are imperfectly complementary and their alignment can generate mismatches and single-strand nicks (Fig. 4A). C-NHEJ is just as efficient with fully complementary ends as with imperfectly complementary ends and is mainly error-free with fully complementary ends.43 With imperfectly complementary ends, C-NHEJ can adapt to perform efficient ligation, generating imperfect alignments and limiting genetic instability.43 In conclusion, A-EJ is exclusively error-prone, whereas C-NHEJ (which blocks A-EJ) is mainly error-free but adaptable for imperfect ends (Fig. 4B). Thus, the structure of the DNA, rather than the accuracy or inaccuracy of the C-NHEJ repair machinery itself, dictates the final outcome.
Figure 4.

The accuracy of end joining. (A) The fidelity of C-NHEJ on fully (left panel) or incompletely (right panel) complementary ends. Here, examples are given with ends generated by the meganuclease I-SceI. The cleavage sites are not palindromic therefore 2 I-SceI cleavage sites in the same orientations yield fully complementary ends (left panel), whereas 2 I-SceI cleavage sites in inverted orientation yield incompletely complementary overhangs (right panel). A-EJ leads to deletions with both types of DNA ends. C-NHEJ is mainly error-free on fully complementary ends and uses 3 classes of imperfect annealing (3 out of the 4 3′ protruding nucleotides generated by I-SceI cleavage) with non-fully complementary ends. Thus, C-NHEJ is conservative but adaptable for incompletely complementary ends. (B) The actual accuracy of end joining. A-EJ is highly mutagenic in all situations but it is blocked by C-NHEJ, which can act on imperfectly complementary ends. In situations producing non-ligatable ends, such as hairpins in V(D)J recombination or IR-induced multiple damages at DSBs, a preliminary processing step is required prior to end joining. Note that in these cases, diversity or mutagenesis is generated by the processing step rather than the end-joining machinery. (C) Chromothripsis. The religation of shattered chromosomes in small pieces (colored squares) leads to a combination of rearranged chromosomes in addition to amplification and loss of the small pieces. (D) A model for chromothripsis arising through MMBIR (microhomology-mediated break induced replication). Resection of the DNA double-strand end generates a 3′ overhang, which can anneal another DNA molecule (in red) through the use of microhomologies (blue squares), thus priming DNA synthesis (dotted arrow). This mechanism can lead to more complex rearrangements if it is coupled to multiple cycles of disassembly and template switches of the replication forks (right panel), still using microhomologies.
The Importance of Being Versatile
Although both HR and NHEJ are essential for the maintenance of genome stability, they are also versatile because they can process imperfectly complementary or homologous DNA. In addition, both repair pathways can promote genetic rearrangements. Organisms take advantage of these capabilities to generate genetic diversity through highly controlled processes. In these cases, DSBs are generated by cell nucleases and DSB repair is performed using the same repair processes as those used for accidental DSBs.
HR and genetic diversity
Because of its versatility, HR is involved in many essential biological processes, ranging from molecular evolution to DNA repair and meiotic differentiation.
During meiosis, HR favors allelic recombination between the 2 homologous chromosomes. Because the 2 homologous chromosomes are not fully identical and the process favors CO, this process ensures mixing of alleles, thus creating genetic diversity.
The complexity of the immune repertoire in chickens is ensured by GC between an expression allele and pseudogenes.44
In pathogens, antigenic variation allows immune evasion from the host. GC between the expressed copy and an archive of silent gene copies is prominently used for antigenic variation.45
HR is a driving force for the evolution of multigene families. In some families of repeated genes, the duplicated genes co-evolved in a phenomenon called concerted evolution.46 GC is the driving force of concerted evolution.
The necessity for NHEJ to adapt to imperfect DNA ends
V(D)J recombination
Genetic diversity at the immunoglobulin gene locus is required for an efficient immune response. This genetic diversity is achieved through several successive processes. Rearrangement of the (V), (D), and (J) segments generates the coding joint and the signal joint. Two processes increase the diversity at the coding joints: (1) Cleavage by Rag1/Rag2 generates hairpins at the broken coding ends, and subsequent hairpin resolution generates a combination of sequences at the ends; (2) The terminal deoxynucleotidyl transferase (TdT) adds non-templated nucleotides (N-) to the coding joints, thus increasing junctional diversity.8 Notably, the diversity is generated through accessory mechanisms (hairpin resolution or TdT) rather than through C-NHEJ itself. The necessity for additional processes to generate diversity at the resealed junction supports the notion that C-NHEJ is not sufficiently mutagenic per se. Moreover, the repair of signal joints, which involves direct C-NHEJ-mediated ligation, is largely error-free. Thus, when the DNA ends are directly suitable for ligation, C-NHEJ is error-free. Notably, the DNA ends at the coding joint are not fully complementary after the action of TdT but because of its versatility C-NHEJ is still able to join these DNA ends. Remarkably, the DNA ends are not complementary during class-switch recombination, and the versatility of C-NHEJ is therefore also essential to complete this process.
DSBs resulting from exposure to ionizing radiation (IR)
The DSBs generated by IR are chemically altered at the DNA ends. Complex lesions are produced that make the DNA ends chemically inept for enzymatic ligation. This situation is different from that of the imperfect complementarity of “biochemically clean” ends because the ligase is inactive on these chemically modified ends. Thus, DSBs generated by IR must first be processed prior to ligation. Mutagenesis at the re-sealed junctions of IR-induced DSBs actually results from this preliminary “cleaning” step, rather than from the putative inaccuracy of the C-NHEJ machinery itself.
The preliminary “cleaning” step generates non-complementary ends. The versatility of C-NHEJ is essential for repair of IR-induced DSBs; otherwise, organisms would be highly sensitive to IR even at low doses. The adaptability of C-NHEJ is thus essential for resistance to IR. This is of crucial importance for the consequences of low-dose exposure to IR, such as environmental or medical (radiological examination) exposures.
The Specific Case of Telomere Protection
Because of the linear structure of chromosomes, their extremities would have exposed double-stranded DNA ends. Ligation of these chromosome ends would generate chromosome fusions, which are detrimental for genetic stability. To prevent such events the chromosome ends are protected by telomeres, which adopt specific structures known as telomeric-loops (“T-Loops”) that prevent inappropriate recognition as DNA double strand breaks by DNA resection enzymes, C-NHEJ machinery, and activating DNA damage pathways. A dedicated complex of 6 factors (TRF1, TRF2, TPP1, POT1, RAP1, and TIN2), called the telosome or “Shelterin” complex, protects these terminal regions by capping them.47
Telomere protection: a dual relationship with DNA repair factors
The telosome is an essential structure that interacts with several factors to ensure the maintenance, protection, and stability of genetic information. The T-Loop structure and Shelterin complex repress 6 different DNA damage response pathways: ATM and ATR kinases, C-NHEJ, A-EJ, HR, and resection. Drastic shortening of the telomeres or defects in telomere regulation lead to dysfunctional telomeres, which aberrantly activate the DNA damage response (DDR) to initiate cell cycle arrest, senescence, or apoptosis.48 Uncapping the telomeres may promote telomere fusion between chromosomes by NHEJ, and the attrition of telomere length promotes problems with end replication. All of these events may be considered markers of genomic instability.49,50
TRF2 blocks ATM signaling whereas POT1 negatively regulates ATR signaling. These repressive events prevent the recognition of dysfunctional telomeres as broken DNA by repair factors, making it impossible to fuse the telomeres by NHEJ. The absence of TRF2 leads to the accumulation of 53BP1 and γ-H2AX, which promotes NHEJ at dysfunctional telomeres.49,50 Moreover, TRF2 interacts with RAP1 as a repressor of C-NHEJ at telomeres and HR signaling.49,50
However, recent studies suggest that DDR activation by dysfunctional telomeres is distinct from the response pathway induced by actual DNA damage,51 and that MRN plays an important role at unprotected telomeres.52 Finally, the importance of RIF1 at telomeres and its ability to promote telomere homeostasis were recently reported.53
DSB Repair Deregulation in Cancer: Cause and Consequence?
Defects in HR result in a predisposition to cancer. Indeed, many breast tumor protection genes are involved in DSB repair.54 In addition, the oncogenic kinase AKT1, which is activated in 40–60% of sporadic breast cancers and 40% of sporadic ovarian cancers, is associated with DSB repair (for a review, see55). Specifically, AKT1 inhibits HR by sequestering BRCA1 and RAD51 in the cytoplasm.41 Furthermore, PTEN, one of the genes mutated in familial breast cancer, is an antagonist of AKT1. PTEN inactivation has been shown to increase genetic instability and to decrease the expression of RAD51. Additionally, Bcl-2 overexpression inhibits HR and NHEJ.56
On the other hand, stimulation of HR has also been reported in cancer:
>p53 represses HR (for a review, see39).
Bloom syndrome (BS) results from inactivation of the BLM protein, a member of the RecQ helicase family that plays an important role in the resolution of HR intermediates, the processing of blocked replication forks, and the initiation of DNA double-strand break repair.9,38 BS cells exhibit increased levels of SCE and hyper-recombination phenotypes.57
The oncogenic BCR/ABL fusion derives from translocation of the cABL gene from chromosome 9 to the BCR gene locus on chromosome 22. This translocation is present in patients with chronic myelogenous leukemia (CML) patients and in many cases of acute lymphocytic leukemia. The BCR/ABL fusion protein exhibits constitutive tyrosine kinase activity. Expression of BCR/ABL increases the intracellular level of the RAD51 protein by various mechanisms58: i) transcriptional activation, ii) inhibition of caspase-3–mediated RAD51 protein degradation, and iii) high levels of constitutive Tyr315 phosphorylation of RAD51 through interaction with BCR/ABL. This phosphorylation and RAD51-dependent HR control resistance to cisplatin and mitomycin C.58 In contrast, BCR/ABL expression inhibits DNA-PK activity, which is involved in NHEJ.59
Genome modifications are a hallmark of cancers, and many of them have been linked to DSB repair. Translocations are highly frequent in tumor cells60 and are proposed to be the cause of 20% of cancers.61 Interestingly, sequencing of translocation junctions revealed the presence of microhomologies, implicating A-EJ.62
Cancer cells are also characterized by insertions/deletions (INDELs) ranging from 1 nt to megabases in length. INDELs represent the second most abundant genome modification after single nucleotide polymorphisms (SNPs). INDELs can result from replication errors, particularly on repeated sequences, or from end joining.
In addition to the classic theory that cancer develops through the accumulation of successive mutations leading to carcinogenesis, recent studies have shown that massive genome rearrangements can also occur on a very short timescale (one cell cycle) in tumor cells.
In chromothripsis, which has been described in a broad range of cancers, the genome is scattered and then reassembled by tens to thousands of genomic rearrangements occurring in one cellular crisis (Fig. 4C).63,64 Examination of these rearrangements has implicated A-EJ because the junction sequences contain microhomology as well as insertions or deletions of variable sizes.65,66 However, this mechanism cannot account for amplification events. Replication-based repair pathways are more plausible mechanisms to account for both genomic gains and losses. A hybrid of replication-independent and replication-dependent processes has been proposed to explain these complex rearrangements: microhomology-mediated break-induced replication (MMBIR) (Fig. 4D),67 which is associated with a specific mechanism linked to replication blocks, fork stalling, and template switching (FoSTeS).68 However, such replication forks are weakly processive and can undergo several rounds of template switching, generating complex rearrangements with deletions, amplifications, and non-reciprocal translocations.
Another mutational phenomenon called kataegis was recently described, in which a number of mutations accumulate during a single cell cycle in hotspots consisting of hundreds of bases to megabases that are very often close to rearrangement breakpoints.69,70 At least for some events, these mutations have been proposed to be a consequence of cytidine deamination events by proteins of the AID/APOBEC family on long ssDNA stretches generated by HR. Overexpression of APOBEC3B correlates with an elevated level of mutations in breast tumors and cell lines.70-73 The occurrence of kataegis on ssDNA stretches generated during HR has also been shown in yeast.73 Other mechanisms leading to ssDNA stretches, such as uncoupled replication forks, are also potential templates for kataegis.70
DSBs: A Weapon Against Cancer Cells
Despite the potential role of DSB repair in tumorigenesis, DSB-inducing agents are often used as anticancer therapies. Extensive research, including preclinical and clinical phase trials, is therefore ongoing to develop new strategies and/or new molecules to potentiate the impact of these agents, particularly through the development of DSB repair or DDR inhibitors.74 At the present time, there are several validated inhibitors targeting NHEJ factors such as DNA-PKcs and DNA ligase IV, and others targeting HR, such as the MRN complex, RPA, and Rad51. Mediators and transducers of DSBs responses, such as ATM, ATR, Chk1/2, and p53, and cell cycle checkpoint regulators (Wee1 and Cdc25) also constitute potentially promising targets.
The concept of synthetic lethality has recently emerged, in which tumor cells deficient in one pathway rely more on other pathways to survive and the latter pathway becomes the target. An ideal example is the use of PARP inhibitors in the treatment of HR-deficient cancer cells (for example, those with BRCA1 or BRCA2 mutations).75,76
Lastly, a new perspective will be to elucidate the link between haploinsufficiency of DDR factors and promotion of carcinogenesis to try to apply the same strategy of synthetic lethality.77
Genome Manipulation Through DSB Repair
Both HR and NHEJ are now widely used for targeted genome engineering. HR allows targeted gene replacement, which can introduce a controlled mutation or correct a mutated endogenous gene. Unfortunately, HR is not a very efficient system and is restricted to the S and G2 phases, and thus to proliferating cells. One solution is to stimulate HR through the generation of a DSB in the target sequence. Several strategies have been developed along these lines using engineered nucleases specific to the target locus, such as meganucleases, zinc-finger nucleases, TAL nucleases, or CRISPR nucleases.78
NHEJ is also used to generate targeted mutagenesis. Following targeted cleavage by a specific nuclease (see above), selection of unfaithful end-joining events allows the isolation of targeted knockout mutants. However, since C-NHEJ is error free this strategy primarily relies on A-EJ–mediated events. This approach raises several concerns:
A-EJ is initiated by uncontrolled resection at the repaired junctions.
A-EJ favors translocations, thus potentially generating the risk of off-target genetic instability.
A-EJ is less efficient than C-NHEJ.43
Because it is a conservative process, strategies based on C-NHEJ should preferably be chosen, provided that controlled variability is introduced at the repair junction. Interestingly, ectopically expressed TdT efficiently generates variability at DNA ends generated by the meganuclease I-SceI in a C-NHEJ-dependent manner.79 Other DNA end-modifying enzymes have been shown to generate mutations at the resealed junctions of DNA ends generated by TAL endonucleases.80 Selection of systems that act through C-NHEJ-dependent pathways should minimize the risk of genomic instability.
Concluding Remarks
Although A-EJ and SSA are necessarily deleterious to genome stability, HR and NHEJ are double-edged swords because they are both necessary for maintenance of genome stability yet can also generate genetic diversity or instability. HR and NHEJ share similarities in their impacts on genome stability:
HR and NHEJ are both necessary for the maintenance of genome stability.
HR and NHEJ are both involved in processes that generate genome diversity.
HR and NHEJ can both generate genome rearrangements.
In both cases, the structure of the DNA molecules dictates the final product.
However, they also exhibit differences:
HR has the intrinsic ability to affect the structure of genetic material in association with the initiation of error-prone DNA synthesis.
NHEJ is not intrinsically error prone, but its adaptability and versatility allow it to act on imperfect ends (for a review see3).
Therefore, the relative contributions of HR and NHEJ to genomic instability during cancer development need to be carefully re-examined.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank everyone who made helpful comments. We apologize to our colleagues whose contributions are not cited due to space constraints.
Funding
This work is supported by funding from the Ligue Nationale Française contre le Cancer and the INCa (Institut National du Cancer).
References
- 1. Haber JE. Genome stability. DNA Repair Recombination. Talor and Francis Group: New York; 2014. [Google Scholar]
- 2. Magdalou I, Lopez BS, Pasero P, Lambert SA. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol 2014; 30C:154-64; http://dx.doi.org/ 10.1016/j.semcdb.2014.04.035 [DOI] [PubMed] [Google Scholar]
- 3. Betermier M, Bertrand P, Lopez BS. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet 2014; 10:e1004086; PMID: 24453986; http://dx.doi.org/ 10.1371/journal.pgen.1004086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 2009; 16:819-24; PMID: 19633668; http://dx.doi.org/ 10.1038/nsmb.1641 [DOI] [PubMed] [Google Scholar]
- 5. Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 2001; 15:3237-42; PMID: 11751629; http://dx.doi.org/ 10.1101/gad.946401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. . 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; PMID: 20362325; http://dx.doi.org/ 10.1016/j.cell.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Langerak P, Mejia-Ramirez E, Limbo O, Russell P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet 2011; 7:e1002271; PMID: 21931565; http://dx.doi.org/ 10.1371/journal.pgen.1002271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013; 152:417-29; PMID: 23374339; http://dx.doi.org/ 10.1016/j.cell.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grabarz A, Guirouilh-Barbat J, Barascu A, Pennarun G, Genet D, Rass E, Germann SM, Bertrand P, Hickson ID, Lopez BS. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep 2013; 5:21-8; PMID: 24095737; http://dx.doi.org/ 10.1016/j.celrep.2013.08.034 [DOI] [PubMed] [Google Scholar]
- 10. Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 2013; 49:858-71; PMID: 23333305; http://dx.doi.org/ 10.1016/j.molcel.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. . Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 2013; 339:711-5; PMID: 23306439; http://dx.doi.org/ 10.1126/science.1230624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 2013; 339:700-4; PMID: 23306437; http://dx.doi.org/ 10.1126/science.1231573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. . 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 2013; 153:1266-80; PMID: 23727112; http://dx.doi.org/ 10.1016/j.cell.2013.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jowsey PA, Doherty AJ, Rouse J. Human PTIP facilitates ATM-mediated activation of p53 and promotes cellular resistance to ionizing radiation. J Biol Chem 2004; 279:55562-9; PMID: 15456759; http://dx.doi.org/ 10.1074/jbc.M411021200 [DOI] [PubMed] [Google Scholar]
- 15. Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci 2009; 34:264-72; PMID: 19375328; http://dx.doi.org/ 10.1016/j.tibs.2009.01.010 [DOI] [PubMed] [Google Scholar]
- 16. Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol 2009; 16:808-13; PMID: 19633670; http://dx.doi.org/ 10.1038/nsmb.1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol 2009; 16:814-8; PMID: 19633669; http://dx.doi.org/ 10.1038/nsmb.1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature 2007; 450:509-14; PMID: 17965729; http://dx.doi.org/ 10.1038/nature06337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. . DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 2014; 53:7-18; PMID: 24316220; http://dx.doi.org/ 10.1016/j.molcel.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol 2004; 24:9478-86; PMID: 15485915; http://dx.doi.org/ 10.1128/MCB.24.21.9478-9486.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev 2006; 20:1721-6; PMID: 16818604; http://dx.doi.org/ 10.1101/gad.1431006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buis J, Stoneham T, Spehalski E, Ferguson DO. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat Struct Mol Biol 2012; 19:246-52; PMID: 22231403; http://dx.doi.org/ 10.1038/nsmb.2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 2009; 284:9558-65; PMID: 19202191; http://dx.doi.org/ 10.1074/jbc.M808906200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, Burma S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun 2014; 5:3561; PMID: 24705021; http://dx.doi.org/ 10.1038/ncomms4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caestecker KW, Van de Walle GR. The role of BRCA1 in DNA double-strand repair: past and present. Exp Cell Res 2013; 319:575-87; PMID: 23200932; http://dx.doi.org/ 10.1016/j.yexcr.2012.11.013 [DOI] [PubMed] [Google Scholar]
- 26. Jiang G, Plo I, Wang T, Rahman M, Cho JH, Yang E, Lopez BS, Xia F. BRCA1-Ku80 interaction enhances end-joining fidelity of chromosomal double-strand breaks in G1 phase of the cell cycle. J Biol Chem 2013; 288:8966-76; PMID: 23344954; 18923075http://dx.doi.org/10.1074/jbc.M112.412650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 2008; 22:2767-72; PMID: 18923075; http://dx.doi.org/ 10.1101/gad.503108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A 2008; 105:16906-11; PMID: 18971343; http://dx.doi.org/ 10.1073/pnas.0809380105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guirouilh-Barbat J, Lambert S, Bertrand P, Lopez BS. Is homologous recombination really an error-free process? Front Genet 2014; 5:175; PMID: 24966870; http://dx.doi.org/ 10.3389/fgene.2014.00175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol 2010; 17:410-6; PMID: 20208544; http://dx.doi.org/ 10.1038/nsmb.1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, et al. . DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet 2011; 7:e1002080; PMID: 21655080; http://dx.doi.org/ 10.1371/journal.pgen.1002080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilhelm T, Magdalou I, Barascu A, Techer H, Debatisse M, Lopez BS. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc Natl Acad Sci U S A 2014; 111:763-8; PMID: 24347643; http://dx.doi.org/ 10.1073/pnas.1311520111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014; 343:88-91; PMID: 24310611; http://dx.doi.org/ 10.1126/science.1243211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 2010; 329:82-5; PMID: 20595613; http://dx.doi.org/ 10.1126/science.1191125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A. Break-induced replication is highly inaccurate. PLoS Biol 2011; 9:e1000594; PMID: 21347245; http://dx.doi.org/ 10.1371/journal.pbio.1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Iraqui I, Chekkal Y, Jmari N, Pietrobon V, Freon K, Costes A, Lambert SA. Recovery of arrested replication forks by homologous recombination is error-prone. PLoS Genet 2012; 8:e1002976; PMID: 23093942; http://dx.doi.org/ 10.1371/journal.pgen.1002976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet 2000; 25:192-4; PMID: 10835635; http://dx.doi.org/ 10.1038/76055 [DOI] [PubMed] [Google Scholar]
- 38. Bernstein KA, Gangloff S, Rothstein R. The RecQ DNA helicases in DNA repair. Annu Rev Genet 2010; 44:393-417; PMID: 21047263; http://dx.doi.org/ 10.1146/annurev-genet-102209-163602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bertrand P, Saintigny Y, Lopez BS. p53's double life: transactivation-independent repression of homologous recombination. Trends Genet 2004; 20:235-43; PMID: 15145576; http://dx.doi.org/ 10.1016/j.tig.2004.04.003 [DOI] [PubMed] [Google Scholar]
- 40. Laulier C, Barascu A, Guirouilh-Barbat J, Pennarun G, Le Chalony C, Chevalier F, Palierne G, Bertrand P, Verbavatz JM, Lopez BS. Bcl-2 inhibits nuclear homologous recombination by localizing BRCA1 to the endomembranes. Cancer Res 2011; 71:3590-602; PMID: 21444675; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3119 [DOI] [PubMed] [Google Scholar]
- 41. Plo I, Laulier C, Gauthier L, Lebrun F, Calvo F, Lopez BS. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res 2008; 68:9404-12; PMID: 19010915; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0861 [DOI] [PubMed] [Google Scholar]
- 42. Sundararajan R, Gellon L, Zunder RM, Freudenreich CH. Double-strand break repair pathways protect against CAG/CTG repeat expansions, contractions and repeat-mediated chromosomal fragility in Saccharomyces cerevisiae. Genetics 2010; 184:65-77; PMID: 19901069; http://dx.doi.org/ 10.1534/genetics.109.111039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, Lopez BS. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell 2004; 14:611-23; PMID: 15175156; http://dx.doi.org/ 10.1016/j.molcel.2004.05.008 [DOI] [PubMed] [Google Scholar]
- 44. Reynaud CA, Anquez V, Grimal H, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell 1987; 48:379-88; PMID: 3100050; http://dx.doi.org/ 10.1016/0092-8674(87)90189-9 [DOI] [PubMed] [Google Scholar]
- 45. Palmer GH, Brayton KA. Antigenic variation and transmission fitness as drivers of bacterial strain structure. Cell Microbiol 2013; 15:1969-75; PMID: 23941262; http://dx.doi.org/ 10.1111/cmi.12182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arnheim N. Concerted evolution of multigene families. In: Evolution of genes and proteins. Koehn RK. and Nei M. (Ed). Sinauer Associates: Sunderland, MA, 1983; 38-61 [Google Scholar]
- 47. Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet 2008; 42:301-34; PMID: 18680434; http://dx.doi.org/ 10.1146/annurev.genet.41.110306.130350 [DOI] [PubMed] [Google Scholar]
- 48. d’Adda di Fagagna F, Teo SH, Jackson SP. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev 2004; 18:1781-99; http://dx.doi.org/ 10.1101/gad.1214504 [DOI] [PubMed] [Google Scholar]
- 49. O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 2010; 11:171-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science 2012; 336:593-7; PMID: 22556254; http://dx.doi.org/ 10.1126/science.1218498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cesare AJ, Hayashi MT, Crabbe L, Karlseder J. The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol Cell 2013; 51:141-55; PMID: 23850488; http://dx.doi.org/ 10.1016/j.molcel.2013.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deng Y, Guo X, Ferguson DO, Chang S. Multiple roles for MRE11 at uncapped telomeres. Nature 2009; 460:914-8; PMID: 19633651; http://dx.doi.org/ 10.1038/nature08196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shi T, Bunker RD, Mattarocci S, Ribeyre C, Faty M, Gut H, Scrima A, Rass U, Rubin SM, Shore D,et al. Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell 2013; 153:1340-53; PMID: 23746845; http://dx.doi.org/ 10.1016/j.cell.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 54. Filippini SE, Vega A. Breast cancer genes: beyond BRCA1 and BRCA2. Front Biosci (Landmark Ed) 2013; 18:1358-72; PMID: 23747889; http://dx.doi.org/ 10.2741/4185 [DOI] [PubMed] [Google Scholar]
- 55. Guirouilh-Barbat J, Wilhelm T, Lopez BS. AKT1/BRCA1 in the control of homologous recombination and genetic stability: the missing link between hereditary and sporadic breast cancers. Oncotarget 2010; 1:691-9; PMID: 21321378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Laulier C, Lopez BS. The secret life of Bcl-2: Apoptosis-independent inhibition of DNA repair by Bcl-2 family members. Mutat Res 2012; 751:247-57; PMID:22677530 [DOI] [PubMed] [Google Scholar]
- 57. Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer 2009; 9:644-54; PMID: 19657341; http://dx.doi.org/ 10.1038/nrc2682 [DOI] [PubMed] [Google Scholar]
- 58. Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G, Nowicki MO, Pierce AJ, Fishel R, Skorski T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell 2001; 8:795-806; PMID: 11684015; http://dx.doi.org/ 10.1016/S1097-2765(01)00357-4 [DOI] [PubMed] [Google Scholar]
- 59. Deutsch E, Dugray A, AbdulKarim B, Marangoni E, Maggiorella L, Vaganay S, M’Kacher R, Rasy SD, Eschwege F, Vainchenker W, et al. . BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001; 97:2084-90; PMID: 11264175; http://dx.doi.org/ 10.1182/blood.V97.7.2084 [DOI] [PubMed] [Google Scholar]
- 60. Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer 2013; 13:443-54; PMID: 23760025; http://dx.doi.org/ 10.1038/nrc3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007; 7:233-45; PMID: 17361217; http://dx.doi.org/ 10.1038/nrc2091 [DOI] [PubMed] [Google Scholar]
- 62. Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, Quinlan AR, Hall IM. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res 2013; 23:762-76; PMID: 23410887; http://dx.doi.org/ 10.1101/gr.143677.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. . The genomic complexity of primary human prostate cancer. Nature 2011; 470:214-20; PMID: 21307934; http://dx.doi.org/ 10.1038/nature09744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. . Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144:27-40; PMID: 21215367; http://dx.doi.org/ 10.1016/j.cell.2010.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, Jager N, Remke M, Shih D, Northcott PA, et al. . Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012; 148:59-71; PMID: 22265402; http://dx.doi.org/ 10.1016/j.cell.2011.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. . The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486:400-4; PMID: 22722201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 2009; 5:e1000327; PMID: 19180184; http://dx.doi.org/ 10.1371/journal.pgen.1000327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 2009; 41:849-53; PMID: 19543269; http://dx.doi.org/ 10.1038/ng.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. . Mutational processes molding the genomes of 21 breast cancers. Cell 2012; 149:979-93; PMID: 22608084; http://dx.doi.org/ 10.1016/j.cell.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. . Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell 2012; 46:424-35; PMID: 22607975; http://dx.doi.org/ 10.1016/j.molcel.2012.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, et al. . APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013; 494:366-70; PMID: 23389445; http://dx.doi.org/ 10.1038/nature11881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chan K, Sterling JF, Roberts SA, Bhagwat AS, Resnick MA, Gordenin DA. Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet 2012; 8:e1003149; PMID: 23271983; http://dx.doi.org/ 10.1371/journal.pgen.1003149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. Elife 2013; 2:e00534; PMID: 23599896; http://dx.doi.org/ 10.7554/eLife.00534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jekimovs C, Bolderson E, Suraweera A, Adams M, O’Byrne KJ, Richard DJ. Chemotherapeutic Compounds Targeting the DNA Double-Strand Break Repair Pathways: The Good, the Bad, and the Promising. Front Oncol 2014; 4:86; PMID: 24795863; http://dx.doi.org/ 10.3389/fonc.2014.00086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434:913-7; PMID: 15829966; http://dx.doi.org/ 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- 76. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. . Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917-21; PMID: 15829967; http://dx.doi.org/ 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- 77. Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26:7773-9; PMID: 18066090; http://dx.doi.org/ 10.1038/sj.onc.1210881 [DOI] [PubMed] [Google Scholar]
- 78. Gaj T, Gersbach CA, Barbas CFr. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013; 31:397-405; PMID: 23664777; http://dx.doi.org/ 10.1016/j.tibtech.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Boubakour-Azzouz I, Bertrand P, Claes A, Lopez BS, Rougeon F. Terminal deoxynucleotidyl transferase requires KU80 and XRCC4 to promote N-addition at non-V(D)J chromosomal breaks in non-lymphoid cells. Nucleic Acids Res 2012; 40:8381-91; PMID: 22740656; http://dx.doi.org/ 10.1093/nar/gks585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Certo MT, Gwiazda KS, Kuhar R, Sather B, Curinga G, Mandt T, Brault M, Lambert AR, Baxter SK, Jacoby K, et al. . Coupling endonucleases with DNA end-processing enzymes to drive gene disruption. Nat Methods 2012; 9:973-5; PMID: 22941364; http://dx.doi.org/ 10.1038/nmeth.2177 [DOI] [PMC free article] [PubMed] [Google Scholar]