

Abstract
DYRK1A (dual-specificity tyrosine-regulated kinase 1A) is a kinase with multiple implications for embryonic development, especially in the nervous system where it regulates the balance between proliferation and differentiation of neural progenitors. The DYRK1A gene is located in the Down syndrome critical region and may play a significant role in the developmental brain defects, early neurodegeneration, and cancer susceptibility of individuals with this syndrome. DYRK1A is also expressed in adults, where it might participate in the regulation of cell cycle, survival, and tumorigenesis, thus representing a potential therapeutic target for certain types of cancer. However, the final readout of DYRK1A overexpression or inhibition depends strongly on the cellular context, as it has both tumor suppressor and oncogenic activities. Here, we will discuss the functions and substrates of DYRK1A associated with the control of cell growth and tumorigenesis with a focus on the potential use of DYRK1A inhibitors in cancer therapy.
Keywords: cancer, cell proliferation, cell differentiation, DYRK1A, neural progenitors
Introduction
DYRK (dual-specificity tyrosine-regulated kinase) family members represent a subfamily of protein kinases that have been identified in distantly related organisms such as yeast, Drosophila, and human. Seven mammalian Dyrk-related kinases have been identified: DYRK1A, DYRK1B, DYRK1C, DYRK2, DYRK3, DYRK4A, and DYRK4B.1,2 The DYRK proteins are dual-specificity protein kinases that autophosphorylate a conserved tyrosine (Y) residue in their own activation loop but phosphorylate their substrates at serine (S) or threonine (T) residues.3,4 The Y autophosphorylation occurs during translation and induces kinase activation; however, once the protein is fully translated, kinase activity becomes restricted to S and T residues and no longer depends on Y phosphorylation.5,6
DYRK1A is the most extensively studied among this family of kinases because its gene maps to human chromosome 21 within the Down syndrome critical region (DSCR).7-9 Moreover, this kinase is overexpressed in the brain of patients with Down syndrome (DS) and many of its known substrates have been linked to neuropathologic traits of this syndrome.10,11 In fact, accumulating evidence in experimental models suggests that DYRK1A inhibitors can reverse some of the neurologic alterations associated with its overexpression and therefore could be of use in individuals with DS.12 These aspects have been extensively reviewed elsewhere. However, there are other less well-known facets of DYRK1A, such as its participation in cancer. Many of its downstream targets are associated with the control of cell growth and survival, especially in the nervous system, where it has been mostly studied, but also in other tissues. Interestingly, in the cancer context, DYRK1A activity might be linked to both oncogenesis and tumor suppression. Here, we will try to summarize all known implications of DYRK1A function in cell growth and cancer with the aim of understanding this dichotomy, which could have clinically relevant implications for the development of therapeutic DYRK1A inhibitors.
Role of DYRK1A in Embryonic and Adult Neurogenesis
Vertebrate Dyrk1A is expressed ubiquitously in a broad spectrum of embryonic tissues at different stages of development but also in some adult tissues, most prevalently in heart, lung, brain, and skeletal muscle.7-9,13 In mice, the absence of Dyrk1A is lethal at the embryonic stage. Heterozygous animals are viable but have a reduced size at birth that is maintained through adulthood. This reduction is more noticeable in organs such as the brain and liver. Heterozygous mice show decreased neonatal viability, a reduced number of neurons in brain areas, alterations in motor and development, dopaminergic deficiency, and impairment in spatial learning development.14,15 Conversely, transgenic mice overexpressing Dyrk1A also present a neurodevelopmental delay and motor and cognitive deficits.16-18 These data reflect the extreme gene dosage sensitivity of this protein and its relevance during neural system (NS) development, where it controls proliferation, neurogenesis, neural differentiation, cell death, and synaptic plasticity.19,20
Drosophila Dyrk1A mutants (minibrain, mnb-/- flies) develop a smaller adult brain (especially the optic lobes and the central brain hemispheres), which appears to be caused by altered neuroblast proliferation during postembryonic neurogenesis.21 Interestingly, recent evidence suggests that this function is not restricted to the NS as Mnb is also required for normal leg and wing growth control.22 Moreover, truncation of the human DYRK1A gene causes microcephaly,23 further supporting an evolutionary conserved function of this kinase during brain development. There appears to be a dynamic spatiotemporal expression pattern of Dyrk1A that is tightly controlled during vertebrate NS development. There is a transient peak of expression of mouse Dyrk1A immediately before the transition from proliferating to neurogenic divisions.24 After that, its expression is maintained in neural progenitors (NPs), although at a lower level. Later on, Dyrk1A is upregulated in newborn postmitotic neurons and downregulated as the neuron begins to migrate away from the ventricular zone. Once the migrating neuron reaches its target position, Dyrk1A is again expressed before the final differentiation and dendrite formation occurs.25,26 These changes in Dyrk1A expression reinforce the notion that it works as an inhibitor of cell cycle progression. In fact, in utero electroporation of Dyrk1A in the embryonic mouse neocortex inhibits cell proliferation by inducing the nuclear export and degradation of cyclin D1.27 A more recent study indicates that DyrkA kinase activity is responsible for the stabilization of cellular cyclin D1 and the degradation of p27 (a cyclin-dependent kinase [CDK] inhibitor) in mouse and human cells.28 Other authors have shown that upregulation of Dyrk1A induces proliferation arrest of embryonic NPs. Conversely, its loss of function causes overproliferation and cell death in the embryonic chick spinal cord and mouse telencephalon.29 These authors suggest that Dyrk1A is both necessary and sufficient for transcriptional upregulation of the expression of p27. Furthermore, Dyrk1A phosphorylates p53 in rat embryonic hippocampal progenitors H19–7 cells, which leads to a robust induction of p21.30 These results support a model in which Dyrk1A impairs cell cycle progression of embryonic progenitors, especially at the transition from G1/G0 to S phase (summarized in Fig. 1). In contrast with these data, mnb overexpression promotes organ growth through inhibition of the Salvador-Warts-Hippo (SWH) pathway, also called the Hippo pathway,22 a known inhibitor of proliferation and inducer of apoptosis in flies and mammals.31-34 Whether these differences are species- or tissue-specific is not known, but throughout the text we will see more examples of different, and even opposite, readouts of Dyrk1A functions in different contexts. Although there is no clear explanation for this behavior this gene is extremely dosage dependent so one could hypothesize that changes in the level or the duration of Dyrk1A expression could have different consequences. There are even cases in which both downregulation and overexpression of DYRK1A have the same readout.35 Moreover, a recent study using single-cell image analysis has shown that Dyrk1A mediates a dose-dependent increase in the duration of the G1 phase via direct phosphorylation and subsequent degradation of cyclinD1, directing PC12 cells into a reversible arrested state. In contrast, knockdown or kinase inhibition of Dyrk1A greatly increased cyclinD1 protein levels and split cells into 2 fates, with one subpopulation (with low p21 expression) shortening the G1 phase and the other (with high p21 expression) entering a persistent arrested state that differs from the normal quiescence state in which expression of both proteins is low.36 Thus, both upregulation and downregulation of Dyrk1A levels could lead to cell cycle exit (transient or irreversible, respectively) and have similar consequences on tissue growth.
Figure 1.
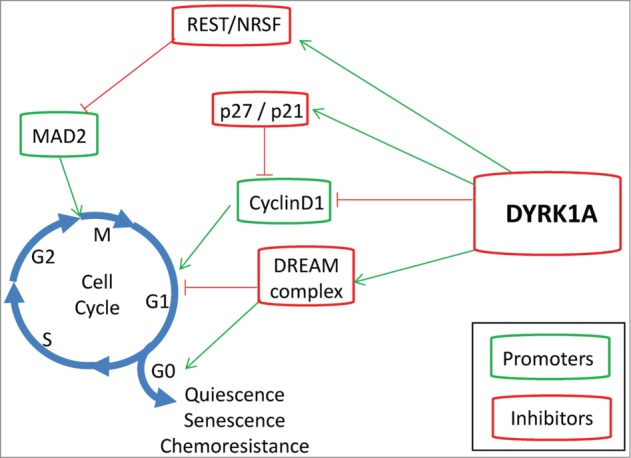
DYRK1A activity blocks cell cycle progression. DYRK1A is considered primarily an inhibitor of proliferation due to its capacity to either block (red lines) cell cycle promoters (green boxes), or activate (green arrows) cell cycle inhibitors (red boxes).
In addition to directly controlling the cell cycle machinery, it has been suggested that overexpression of Dyrk1A is necessary to induce neural differentiation, although it is not clear whether it is sufficient for this final outcome.27,29 Regarding the mechanism, Yang and coworkers have shown that Dyrk1A activity is induced during in vitro differentiation of hippocampal progenitor cells, leading to the stimulation of cAMP responsive element binding protein (CREB) transcriptional activity.37 Another group has suggested that Dyrk1A overexpression potentiates nerve growth factor (NGF)-mediated PC12 neuronal differentiation by upregulating the Ras/MAP kinase signaling pathway.38 More recently, it has been proposed that Dyrk1A phosphorylates Notch in the nuclear compartment and is co-expressed with the Notch ligand Delta1 in single NPs. Furthermore, Dyrk1A suppresses Notch signaling and reduces its capacity to sustain transcription in neural cells, counteracting its antidifferentiative actions.29,39 Another interesting possibility would be that Dyrk1A influences neuronal differentiation through the modulation of RE1-silencing transcription factor (REST, also known as neuron-restrictive silencer factor or NRSF). REST is a zinc finger transcription factor that silences a range of neuronal genes in differentiated non-neuronal tissues and NPs.40 Furthermore, the dissociation of REST and its co-repressors from the RE1 sites is both necessary and sufficient to trigger the transition from pluripotent embryonic stem cells to NPs, and from these to mature neurons.41 Moreover, REST is degraded in the G2 phase and this is necessary for the derepression of Mad2, an essential component of the spindle assembly checkpoint.42 Reduced expression of REST has been observed in cultured fetal DS brain cell-derived neurospheres43 as well as in the brains of DS mouse models.44,45 By inhibiting REST expression, DYRK1A might inhibit the G2-M checkpoint (Fig. 1) as well as promote some of the neural differentiation defects observed in DS.
In contrast to the antiproliferative and prodifferentiative capacity of Dyrk1A during CNS development, we recently suggested that this kinase sustains adult NP self-renewal.46 In our experience, Dyrk1A protein is actively distributed during adult NP cell division and the inherited kinase acts as an inhibitor of epidermal growth factor receptor (EGFR) degradation by phosphorylating Sprouty2 (Spry2), modulator of receptor tyrosine kinases (RTK) turnover. Interestingly DYRK1A phosphorylates T75 on Spry2 and impairs its inhibitory activity on extracellular signal-regulated kinase (ERK) signaling downstream of fibroblast growth factor (FGF).47 However, Spry2 also phosphorylates and sequesters Cbl, a major effector of EGFR degradation.48 The data on adult progenitors suggest that Dyrk1A phosphorylation could be beneficial for the positive function of Spry2 downstream of EGFR but counteractive for the inhibitory function of this protein on FGF signaling (Fig. 3). Therefore, high expression of Dyrk1A should correlate with general activation of RTK function. It would be interesting to test whether phosphorylation by Dyrk1A modulates the binding of Spry2 to regulatory proteins, as it has been described for phosphorylation of other Spry2 residues.49 In fact, Dyrk1A overexpression decreases the interaction of Spry2 and growth factor receptor-bound protein 2 (Grb2), an adaptor between RTK and the ERK pathway.50 These results suggest that DYRK1A kinase activity could have a positive function in both EGFR and FGFR signaling, at least in the context of the adult CNS. In agreement with this, activation of the AKT pathway, a known transducer of RTK activation, has been observed n several transgenic Dyrk1A mouse models, concomitant with an increase in expression of CCND1 (the gene coding for cyclin D1) in postnatal and adult stages.50,51 Together, these data suggest that Dyrk1A might act mainly as a negative regulator of cell cycle during neural development, but also as a positive regulator of cell proliferation in the adult CNS, probably due to differential expression of downstream substrates or regulatory molecules.
Regulation of Quiescence through the DREAM Complex
An important group of proteins that negatively regulate the cell cycle are the retinoblastoma (RB) tumor suppressor protein and the related family members p107 (RBL1) and p130 (RBL2).52 p130 has been shown to accumulate in G0, when it interacts with E2F4 to repress E2F-dependent gene transcription.53-55 P130 and E2F4 are part of a larger multisubunit protein complex, the mammalian DREAM (dimerization partner, RB-like, E2F, and multivulval class B) complex.56,57 In a recent study, phosphorylation of the DREAM component LIN52 at S28 by DYRK1A was shown to regulate complex formation in G0 and E2F repression.58 Inhibition of DYRK1A activity or a point mutation in LIN52 disrupted DREAM assembly and reduced the ability of tumor cells to enter quiescence and the capacity of fibroblasts to undergo Ras-induced senescence58 (Fig. 1). These authors have shown that overexpression of DYRK1A (but not a kinase-deficient mutant) can inhibit proliferation and colony formation of a panel of tumor cell lines from different tissues.58 Moreover, by phosphorylating DYRK1A, LATS2, a component of the Hippo pathway, could enhance the kinase activity of DYRK1A toward the DREAM subunit LIN52. Intriguingly, the LATS2 locus is physically linked with RB1 on 13q, and this region frequently displays loss of heterozygosity in human cancers.59 Together, these results provide a possible explanation for the proposed tumor suppressor role of DYRK1A, especially at the tumor initiation stages. However, the same mechanism could have a protective role later on in the response to chemotherapy. In fact, a recent report indicates that inhibition of DYRK1A could enhance imatinib-induced cell death in gastrointestinal tumors by favoring apoptosis at the expense of cell quiescence.60
Documented Roles of DYRK1A in Cancer
Epidemiologic studies suggest that individuals with DS have an increased risk of acute megakaryoblastic leukemia (AMLK) and acute lymphoblastic leukemia (ALL) in combination with acquired mutations of the transcription factor globin transcription factor 1 (GATA1).61-64 These GATA1 mutations are localized in exon 2 and provoke the expression of a shorter isoform named GATA1s.61 However, non-DS individuals with germline GATA1 mutations similar to those seen in DS-AMLK have no predisposition to leukemia.63 Moreover, trisomy for the DSCR that includes DYRK1A markedly increases the proliferation of megakaryocytes and is sufficient to cooperate with GATA1 mutations in this mouse model, reinforcing the oncogenic function of DYRK1A in this cell type.65 GATA1 is essential for erythroid and megakaryocytic development. GATA1s retains some of the functions of the wild-type protein although it does not repress oncogenic MYC and the proproliferative E2F.66,67 This could explain its oncogenic action but might also lead to a positive feedback loop as E2F stimulates DYRK1A transcription, further increasing DYRK1A expression.68
In contrast to the increased risk of leukemia, individuals with DS show a considerably reduced incidence of most solid tumors.69,70 It has been suggested that overexpression of DYRK1A, in cooperation with DSCR1 (another gene located in the DS critical region) could diminish angiogenesis through attenuation of vascular endothelial growth factor (VEGF)-calcineurin- nuclear factor of activated T cells (NFAT) signaling in endothelial cells.71,72 NFAT proteins are transcription factors that are activated as a result of a calcium flux released from endoplasmic reticulum stores and from the activation of channels in the plasma membrane. This increase in calcium provokes dephosphorylation of NFAT by the phosphatase calcineurin and its translocation to the nucleus, where it cooperates with other factors and coactivators to promote gene transcription.73 Since the discovery of NFAT proteins 2 decades ago as promoters of interleukin (IL)-2 during the activation of T cells,74 it has become increasingly clear that these proteins are not only expressed in immune cells but are also overexpressed in human solid tumors and hematologic malignancies. In general, activation of the NFAT pathway is considered to be a cancer-promoting event by enhancing proliferation and survival (in T cell lymphoid malignancies), by enhancing metastatic dissemination (in epithelial cancers), and by promoting angiogenesis.73,75 Phosphorylation of NFAT by DYRK1A 71,76 primes it for subsequent phosphorylation by casein kinase 1 (CK1) and glycogen synthase kinase 3 (GSK3), which drives the inactivation and nuclear export of NFAT.77-79 As VEGF is one of the known NFAT targets, DYRK1A overexpression could lead to decreased angiogenesis, thus contributing to the lower incidence of adult solid tumors in DS individuals.72 However, it is important to keep in mind that DS is a multifactorial syndrome and the overexpression of other genes could be responsible for the reduced cancer predisposition. Paradoxically, it has been proposed that DYRK1A also contributes to megakaryocytic malignancies through the inhibition of NFAT, although the possibility that other substrates of the kinase are implicated in the oncogenic response has not been excluded.65 Therefore, it seems that while DYRK1A and NFAT suppress the growth of epithelial and lymphoid tumors in the adult, they could act as megakaryocytic oncogenes in children. This led us to hypothesize that DYRK1A has opposite functions during normal development and carcinogenesis of the NS and blood cells.
These context-dependent functions of NFAT have similarities with other well-known substrates of DYRK1A and reinforce the notion that this kinase can have different, and even antagonistic functions, depending on the cellular context or even the cancer stage. This idea has already been suggested by others, at least in the DS context.80 Figure 2 summarizes some of the DYRK1A substrates, focusing on their pro- or antitumoral roles. For example, NOTCH function is inhibited by DYRK1A phosphorylation, as previously mentioned.29,39 Hyperactivation of the NOTCH pathway has classically been viewed as oncogenic in several cancers (solid tumors and blood cancers) although recent studies have revealed tumor suppressor roles for NOTCH signaling in myeloid malignancies.81 Therefore, DYRK1A inhibition in NOTCH-related cancers could have positive or negative consequences for tumor growth and survival. A similar scenario might exist for the protein REST, which has antitumor properties in epithelial cancers but can function as an oncogene for neural tumors.40 REST was first identified as a cancer-related gene a decade ago from studies in medulloblastoma (MD), a childhood tumor of the cerebellum,82,83 and was later also characterized as an oncogene in neuroblastoma (NB), the most common extracranial solid tumor in children84 and glioblastoma (GBM), a deadly adult brain tumor.85,86 These data suggest that REST controls the maintenance of embryonic and adult NPs and, when deregulated, can be implicated in neural tumors. As we previously mentioned, REST appears to be downregulated in DS brains. However, more recent observations indicate that REST can activate DYRK1A transcription through a NRSE site in the human DYRK1A promoter region. Moreover, REST and Dyrk1A are coordinately expressed during neural development, and DYRK1A imbalance can destabilize REST protein expression and reduce its transcriptional activity.35 Therefore, it seems that in this case the regulation is even more complicated: DYRK1A can work as a positive or a negative modulator of REST expression/activity, which in turn could have oncogenic or tumor suppressor activity depending on the tissue.
Figure 2.

Double-edged regulation of tumorigenesis by DYRK1A. DYRK1A has been associated with protumoral activity (green boxes) by activating (green arrows) known oncogenic proteins (red boxes) or by inhibiting (red lines) tumor suppressors (red boxes). However, an antitumoral capacity of DYRK1A has been also described through its activation of tumor suppressors or its inhibition of oncogenic proteins. To add complexity to DYRK1A function, some of the known substrates of this kinase can have both oncogenic and tumor suppressor activities (green and red boxes), depending on the cellular context and the developmental stage.
Survival- or Apoptosis-Related Substrates of DYRK1A
In contrast to the dual (pro- or antitumoral) function of some of the genes mentioned above, there is a list of prosurvival or antiapoptotic proteins that are activated upon DYRK1A phosphorylation and could be important for carcinogenesis in different tissues (Fig. 3). Signal transducer and activator of transcription (STAT) is a latent transcription factor that transmits signals generated primarily by cell surface receptors into the nucleus. STAT3 is transiently activated in normal cells but is constitutively activated in a wide variety of hematologic malignancies (leukemia, lymphomas, and multiple myelomas) and solid tumors (such as head and neck, breast, lung, gastric, hepatocellular, colorectal, brain, and prostate cancers).87 There is strong evidence suggesting that aberrant STAT3 signaling promotes initiation and progression of cancer by either inhibiting apoptosis or inducing proliferation, angiogenesis, invasion, and metastasis.88 Phosphorylation of STAT3 at position Y705 by Janus kinase (JAK) induces its dimerization and nuclear translocation.89 However, the STAT3 molecule contains a second phosphorylation site, S727, within its C terminus that can be phosphorylated by DYRK1A90 and is necessary to achieve maximal transcriptional activity.89 In fact, recent data suggest that phosphorylation at S727 plays a principal role in regulation of cell survival activity and nuclear translocation of STAT3.91 Nevertheless, inhibition of DYRK1A has not been explored as a way to modulate the growth of STAT3-related tumors. GLI1, a major effector of Sonic hedgehog (SHH) signaling, is another oncogenic transcription factor whose nuclear translocation and function seems to be mediated by DYRK1A phosphorylation, most likely through the retention of GLI1 in the nucleus.92 SHH-GLI signaling regulates tumor growth and survival, as well as metastasis, in a number of tumors.93 However, although STAT3 is a bona fide DYRK1A substrate, the relevance of GLI1 phosphorylation by the kinase and the possible applications of DYRK1A inhibitors in SHH-dependent tumors is still under debate.
DYRK1A function has been linked to negative regulation of the intrinsic apoptotic pathway through the phosphorylation of caspase-9.94,95 Caspase-9 is activated by a variety of apoptotic stimuli that trigger the release of cytochrome c from mitochondria. Once in the cytosol, cytochrome c induces the oligomerization of apoptotic protease activating factor 1 (Apaf-1) and the subsequent recruitment of procaspase-9. These events lead to activation of procaspase-9 by autocatalytic processing and consequently to the activation of effector caspase-3 and caspase-7.96 Activation of caspase-9 is inhibited by phosphorylation at T125, which is catalyzed, among others, by Dyrk1A.94,95 This is important during the development of retina progenitors,95 and in the response of cells to hyperosmotic stress.97 Therefore, the inhibition of DYRK1A would lead to caspase-9 induction and could be exploited to enhance the apoptotic response of cancer cells to chemotherapy. In addition, DYRK1A might promote cell survival through phosphorylation and activation of sirtuin 1 (SIRT1; also known as silent mating type information regulation 2 homolog or NAD (+)-dependent protein deacetylase), which participates in the stress response and cellular metabolism.98 It has been shown that DYRK1A and DYRK3 directly phosphorylate SIRT1 at T522, promoting deacetylation of p53, and, more importantly, that the knockdown of endogenous DYRK1A and DYRK3 sensitizes cells to DNA damage-induced cell death.99 These data, together with the impairment of quiescence mediated by inhibition of the DREAM complex and inhibition of the HIPPO pathway (which induces apoptosis) by DYRK1A, suggest that DYRK1A could be a good therapeutic target to increase cell death in response to both chemo- and radiotherapy in different tumors, as has been suggested by others.100
Despite these prosurvival functions of DYRK1A, and excluding its well-known link to DS-related leukemia, it was not until recently that our group described a clear association of this kinase with tumor growth. Our results suggest that DYRK1A is highly expressed in a subset of GBMs, where it correlates with the expression and genetic amplification of EGFR. In parallel to events in normal NPs, downregulation of DYRK1A leads to an increase in EGFR degradation and therefore to inhibition of the self-renewal capacity of GBM cells.101 Furthermore, overexpression of SPRY2 was able to compensate for the EGFR degradation promoted by DYRK1A inhibition and rescue the self-renewal effect. On top of this, we have shown that genetic or pharmacologic blockade of DYRK1A severely impaired tumor growth in vivo, thus opening the door to the use of DYRK1A inhibitors in glioma therapy, at least for EGFR-dependent tumors.101 EGFR is a well-known oncogene in a variety of tumors and inhibitors of its tyrosine kinase activity are being tested in many of these, including GBM, although the results have not always been always optimal.102 Impaired endocytic downregulation of this receptor is frequently associated with cancer. Indeed, dominant-negative forms of CBL have been identified as oncogenes in human myeloid neoplasms103 and SPRY2 has tumor-promoting activity in colon cancer.104 Therefore, the induction of EGFR degradation mediated by inhibition of DYRK1A could represent an alternative strategy for EGFR-related cancers such as colon, lung, and head and neck tumors.
To summarize this section, there is not a simple answer to the question of whether DYRK1A functions as an oncogene or a tumor suppressor (Fig. 2). In addition, it is important to note that the relevance of some of the targets described above has not been confirmed in a tumor context, or even in cancer cell lines. Moreover, in the most conceivable situations several targets would act in a combinatorial way. Therefore, the final response of a cell to DYRK1A overexpression or inhibition probably depends on the molecular and cellular context of each cancer.
DYRK1A Inhibitors and their Possible Use in Cancer Therapeutics
In recent years there has been increased interest in inhibition of DYRK1A activity for the treatment of the mental impairment associated with several neurodegenerative diseases (including Alzheimer's Disease) and DS.12,105 The fact that this kinase is extremely dosage dependent increases its therapeutic potential as a target. The goal then would be not to inhibit DYRK1A completely but rather to a level comparable with that of healthy conditions, which could reduce the potential side effects of targeting this kinase. Most DYRK1A inhibitors are ATP competitors and act by binding within the DYRK1A kinase domain (direct competitors) or by preventing the functionality of the ATP binding site (indirect competitors).6 They can be classified based on their structure (see 100 for a comprehensive review) or based on their origin: i.e., natural compounds and their derivatives, synthetic inhibitors, and broad spectrum kinase inhibitors that show activity against DYRK1A. Table 1 reviews the most potent DYRK1A inhibitors and their possible implications in cancer. Many of them can inhibit other members of the DYRK family, as well as structurally related kinases such as CDKs (important regulators of cell cycle progression) and CDK-like kinases (CLKs), which participate in pre-mRNA splicing.106 Different modifications of these compounds have been envisioned in order to increase specificity and avoid side effects.
Table 1.
DYRK1A inhibitors. The table shows the IC50 of different classes of DYRK1A inhibitors against DYRK1A, other DYRK proteins, and different kinases (only targets with submicromolar IC50 values are included). Cancer-related studies performed with those inhibitors are also indicated
| Compound name | IC50 (nM) |
Evidence related to cancer | |||
|---|---|---|---|---|---|
| (Chemical class) | DYRK1A | Other targets | therapy | Refs. | |
| Natural inhibitors | Harmine (β-carboline) | 22–400 | DYRK1B (166–300), DYRK2 (900–1,900), DYRK3 (800–1000), DYRK4 (80,000), MAO-A (5), CLK1 (27) | Angiogenesis inhibition; Cytotoxic activity in tumor cell lines in vitro (glioma, leukemia, colon, gastric, liver), and in vivo (glioma) |
101,107,111–115,117 |
| EGCG (Polyphenol) | 40–330 | Vimentin (3), COMT (70) | Clinical trials on cancer: urothelial, bladder, prostate, myeloma, breast, lung | 124,132,133 | |
| Staurosporine (glycosilated indolocarbazole) | 20 | Aurora A (7.2), Aurora B (20), Chk1 (1), Ftl3 (3), HGK (1), Ikkb (0.5), Jak2 (1), KDR (10), SYK (4) | 134 | ||
| Synthetic inhibitors | Lamerallins (Chromenoindole) | 40–5,000 | CDK5 (720->10,000), GSK3 (310->10,000), CDK1/CyclinB, PIM1, CK1 (70–8,000) | Apoptosis induction and multidrug resistance phenotype reversion | 119–121,135 |
| INDY (Benzothiazol) | 200 | DYRK1B (240) | Antiproliferative in gliomas | 101,118 | |
| Meriolins (Pyrimidinylindol/azaindol) | 30 | CDK1 (7–170), CDK2 (3–18), CDK5 (3–170), CDK9 (5.6–18), GSK3 (21–400), CK1 (50–200) | Antiproliferative and proapoptotic effects in tumor cells and glioma xenografts | 122,123 | |
| Variolin B (Pyrimidinylindol/azaindol) | 80 | CDK1 (60), CDK2 (80) CDK5 (90) CDK9 (26) GSK3 (70) CK1 (5) | 122 | ||
| Meridianins (Pyrimidinylindol/azaindol) | 34–900 | CDK5 (680->10,000), CK1 (490->10,000), CLK1 (30–70) | 136,137 | ||
| KHCB19 (dichloroindol) | 55 | CLK1 (20), CLK3 (500) | 138 | ||
| Imidazolone (Leucettamine) | 70–1,000 | CLK1 (15–71), GSK3(21–38) | 139 | ||
| Promiscuous kinase inhibitors | Purlavanol A (Purine) | 300 | CDK2 (30–100), PAK4 (100), SRC (<100 ), CDK2/CyclinA (100) | 107,124 | |
| A-443654 (Pyridine) | <10 | DYRK2, DYRK3 (<100 ), ERK8, RSK1, RSK2, PKBα, PKBβ, S6K1, PKA, ROCK2, PRK2, PKCa,PKD1,MSK1, SmMLCK, SK3b, CDK2-CyclinA, PIM1,2,3,MST2,HIPK2 (<100 ) | Initially identified as a potent Akt inhibitor for use in anticancer research field | 140 | |
| TBB, TBI, DMAT, TDI (Tetrahalo-bicycles) | 100–10,000 | DYRK2 (300–35,000), DYRK3 (2,000–5,000), CK2 (100–600), PIM1 (100–1,000), PIM2 (200–4,000), PIM3 (70–1,000) | 117,141–143 | ||
Among DYRK1A inhibitors, the β-carboline alkaloid known as harmine is currently the most selective and effective, although it also blocks other DYRKs, especially DYRK1B. It functions as an ATP-competitive inhibitor to specifically block the S/T kinase activity.6,107 Harmine is isolated from divergent plant species, including the South American vine Banisteriopsis caapi and the mideastern shrub Peganum harmala. It is a component of ayahuasca, a hallucinogenic brew of plant extracts that has been used for centuries in healing and to cure illnesses, including some types of cancer. However, several active principles could be involved in this action and there is no direct evidence that targeting DYRK1A is the relevant anticancer action of ayahuasca.108 Harmine has been shown to inhibit neovessel formation in vitro and in vivo through the regulation of several angiogenic factors and inflammatory cytokines.109 Moreover, harmine and related β-carboniles display cytostatic and/or cytotoxic activity toward cancer cells, including leukemia, colon, liver, gastric, and glioma cells.65,101,110-114 However, harmine has long been known to be a potent inhibitor of monoamine oxidase-A115 and to have hallucinogenic properties as a result of its affinity for the tryptamine and serotonin receptor binding sites.116 These properties seriously limit the in vivo therapeutic applications of this compound. Nonetheless, molecular docking analysis showed that harmine has many degrees of freedom in the ATP-binding pocket of DYRK1A and this could be exploited to more selectively inhibit the kinase.117 Glioma cells are also sensitive in vitro to INDY, a benzothiazol derivative.101 The in vitro biological activity of INDY has been confirmed to involve blockage of Tau phosphorylation and restoration of NFAT activity, and it can rescue the developmental defects of Dyrk1A overexpressing tadpoles in vivo.105,118 However, nothing is currently known about the pharmacokinetic properties of this compound in whole animals or humans, which hampers its therapeutic use. Another 2 synthetic compounds, lamerallins and meriolins, have shown some anticancer and proapoptotic effects in cancer cells and mouse glioma models, although other kinases (especially CDKs) could be implicated in this activity.119-123
Epigallocatechin gallate (EGCG) is a polyphenol and a major catechin component of green tea that shows selective inhibition of DYRK1A compared to other structurally and functionally related kinases by functioning as a noncompetitive inhibitor of ATP.124 Interestingly, a diet rich in green tea clearly improved the brain structure defects and cognitive impairments of DYRK1A-overexpressing mice.51 EGCG has been shown to have anticancer activity by inhibiting topoisomerases I/II, the antiapoptotic enzyme Bcl-xl, and cancer promoting proteases. Moreover, it has additional health properties as it shows potent antibacterial and antiviral activity and elicits antioxidative and anti-inflammatory capacity by suppressing the nitric oxide synthase pathway.105 Although it seems clear that the anticancer activities of green tea and EGCG are not limited to DYRK1A inhibition, their safety for human consumption make them good candidates for use in DYRK1A-related tumors and neurodegenerative diseases.
Most of the inhibitors mentioned in Table 1 have the capacity to block other DYRK proteins, especially the closely related DYRK1B (also known as MIRK). MIRK has been characterized as a negative regulator of cell proliferation that modulates the protein stability of several cell cycle-related molecules.125,126 However, upregulation of MIRK expression and/or constitutive activation of this kinase has been observed in several different types of cancer, where it has been associated with the survival of tumor cells in response to stress.127 For example, depletion of Mirk has been associated with apoptosis in lung cancer128 and also in pancreatic and ovarian cancer cell lines, where it seems to modulate the levels of reactive oxygen species (ROS).129-131 Therefore, the effect of some DYRK1A inhibitors in cancer could also be due to MIRK inhibition, especially if they are associated with changes in the response of tumor cells to hypoxia, nutrient deprivation, and/or cytotoxic stimuli.
Conclusions and Future Perspectives
The results of several in vitro and in vivo studies link DYRK1A activity with cell cycle exit, oncogene-induced senescence, and cell differentiation, especially in the embryonic NS. This notion is reinforced by the nature of many known DYRK1A substrates, and also by the decreased cancer susceptibility of individuals with DS. Therefore one would expect this gene to behave as a tumor suppressor, at least for the initial cancer stages. However, recent evidence indicates that DYRK1A can induce clonogenic and prosurvival properties in certain types of cell or in certain developmental conditions, and that this kinase can be considered as an oncogene for at least 2 types of cancer: myeloid leukemias and gliomas. As a proof of principle, harmine, a potent DYRK1A inhibitor, can block cell growth and tumorigenesis in those tumors. Moreover, DYRK1A might confer chemo- and radioresistance capacities in several tumors by controlling the balance between quiescence and apoptosis. We propose that newly identified DYRK1A inhibitors, which are being developed primarily for their use in mental diseases, should also be considered as anticancer agents, at least for AMKL and GBM. Moreover, considering the relevance of DYRK1A in the NS, it would be important to test the efficacy of such inhibitors in other neural tumors such as oligodendrogliomas, which express high levels of DYRK1A, and MB, given the important cerebellar expression of this kinase. In such brain tumors, the regulation of EGFR turnover by DYRK1A might participate in the oncogenic action of this kinase, in addition to modulation of REST, which is highly expressed in cerebellar NPs, and SHH and/or NOTCH signaling, which are associated with NS development. Special attention should be given to secondary hits of DYRK1A inhibitors and to the fact that DYRK1A blockade could affect several pathways. However, in the case of cancer some promiscuity may be acceptable and might even represent an advantage, further encouraging the preclinical and clinical analysis of such compounds in cancer therapeutics.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by grants from Ministerio de Economía y Competitividad, Fondo de Investigación Sanitaria, PI12/00775 and from Ministerio de Economía y Competitividad, Red Temática de Investigación Cooperativa en Cancer (RD12/0036/0027) to PSG.
References
- 1. Becker W, Joost HG. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog Nucleic Acid Res Mol Biol 1999; 62:1-17; PMID:9932450; http://dx.doi.org/ 10.1016/S0079-6603(08)60503-6 [DOI] [PubMed] [Google Scholar]
- 2. Aranda S, Laguna A, de la LS. DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. FASEB J 2011; 25(2):449-62; PMID:21048044; http://dx.doi.org/ 10.1096/fj.10-165837 [DOI] [PubMed] [Google Scholar]
- 3. Kentrup H, Becker W, Heukelbach J, Wilmes A, Schurmann A, Huppertz C, Kainulainen H, Joost HG. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J Biol Chem 1996; 271(7):3488-95; PMID:8631952; http://dx.doi.org/ 10.1074/jbc.271.7.3488 [DOI] [PubMed] [Google Scholar]
- 4. Himpel S, Panzer P, Eirmbter K, Czajkowska H, Sayed M, Packman LC, Blundell T, Kentrup H, Grötzinger J, Joost HG, et al. . Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem J 2001; 359(Pt 3):497-505; PMID:11672423; http://dx.doi.org/ 10.1042/0264-6021:3590497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lochhead PA, Sibbet G, Morrice N, Cleghon V. Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 2005; 121(6):925-36; PMID:15960979; http://dx.doi.org/ 10.1016/j.cell.2005.03.034 [DOI] [PubMed] [Google Scholar]
- 6. Becker W, Sippl W. Activation, regulation, and inhibition of DYRK1A. FEBS J 2011; 278(2):246-56; PMID:21126318; http://dx.doi.org/ 10.1111/j.1742-4658.2010.07956.x [DOI] [PubMed] [Google Scholar]
- 7. Guimera J, Casas C, Pucharcos C, Solans A, Domenech A, Planas AM, Ashley J, Lovett M, Estivill X, Pritchard MA, et al. . A human homologue of Drosophila minibrain (MNB) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Hum Mol Genet 1996; 5(9):1305-10; PMID:8872470; http://dx.doi.org/ 10.1093/hmg/5.9.1305 [DOI] [PubMed] [Google Scholar]
- 8. Song WJ, Sternberg LR, Kasten-Sportes C, Keuren ML, Chung SH, Slack AC, Miller DE, Glover TW, Chiang PW, Lou L, et al. . Isolation of human and murine homologues of the Drosophila minibrain gene: human homologue maps to 21q22.2 in the Down syndrome "critical region". Genomics 1996; 38(3):331-9; PMID:8975710; http://dx.doi.org/ 10.1006/geno.1996.0636 [DOI] [PubMed] [Google Scholar]
- 9. Shindoh N, Kudoh J, Maeda H, Yamaki A, Minoshima S, Shimizu Y, Shimizu N. Cloning of a human homolog of the Drosophila minibrain/rat Dyrk gene from "the Down syndrome critical region" of chromosome 21. Biochem Biophys Res Commun 1996; 225(1):92-9; PMID:8769099; http://dx.doi.org/ 10.1006/bbrc.1996.1135 [DOI] [PubMed] [Google Scholar]
- 10. Hammerle B, Elizalde C, Galceran J, Becker W, Tejedor FJ. The MNB/DYRK1A protein kinase: neurobiological functions and Down syndrome implications. J Neural Transm Suppl 2003;(67):129-37; PMID:15068245; http://dx.doi.org/ 10.1007/978-3-7091-6721-2_11 [DOI] [PubMed] [Google Scholar]
- 11. Park J, Song WJ, Chung KC. Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell Mol Life Sci 2009; 66(20):3235-40; PMID:19685005; http://dx.doi.org/ 10.1007/s00018-009-0123-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Becker W, Soppa U, Tejedor FJ. DYRK1A: a potential drug target for multiple Down syndrome neuropathologies. CNS Neurol Disord Drug Targets 2014; 13(1):26-33; PMID:24152332; http://dx.doi.org/ 10.2174/18715273113126660186 [DOI] [PubMed] [Google Scholar]
- 13. Okui M, Ide T, Morita K, Funakoshi E, Ito F, Ogita K, Yoneda Y, Kudoh J, Shimizu N. High-level expression of the Mnb/Dyrk1A gene in brain and heart during rat early development. Genomics 1999; 62(2):165-71; PMID:10610708; http://dx.doi.org/ 10.1006/geno.1999.5998 [DOI] [PubMed] [Google Scholar]
- 14. Fotaki V, Dierssen M, Alcantara S, Martinez S, Marti E, Casas C, Visa J, Soriano E, Estivill X, Arbonés ML. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol 2002; 22(18):6636-47; PMID:12192061; http://dx.doi.org/ 10.1128/MCB.22.18.6636-6647.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fotaki V, Martinez dL, Estivill X, Arbones M, Dierssen M. Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav Neurosci 2004; 118(4):815-21; PMID:15301607; http://dx.doi.org/ 10.1037/0735-7044.118.4.815 [DOI] [PubMed] [Google Scholar]
- 16. Smith DJ, Stevens ME, Sudanagunta SP, Bronson RT, Makhinson M, Watabe AM, O'Dell TJ, Fung J, Weier HU, Cheng JF, et al. . Functional screening of 2 Mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with Down syndrome. Nat Genet 1997; 16(1):28-36; PMID:9140392; http://dx.doi.org/ 10.1038/ng0597-28 [DOI] [PubMed] [Google Scholar]
- 17. Ahn KJ, Jeong HK, Choi HS, Ryoo SR, Kim YJ, Goo JS, Choi SY, Han JS, Ha I, Song WJ. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol Dis 2006; 22(3):463-72; PMID:16455265; http://dx.doi.org/ 10.1016/j.nbd.2005.12.006 [DOI] [PubMed] [Google Scholar]
- 18. Altafaj X, Dierssen M, Baamonde C, Marti E, Visa J, Guimera J, Oset M, González JR, Flórez J, Fillat C, et al. . Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum Mol Genet 2001; 10(18):1915-23; PMID:11555628; http://dx.doi.org/ 10.1093/hmg/10.18.1915 [DOI] [PubMed] [Google Scholar]
- 19. Tejedor FJ, Hammerle B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J 2011; 278(2):223-35; PMID:21156027; http://dx.doi.org/ 10.1111/j.1742-4658.2010.07954.x [DOI] [PubMed] [Google Scholar]
- 20. Dierssen M, de Lagran MM. DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A): a gene with dosage effect during development and neurogenesis. ScientificWorldJournal 2006; 6:1911-22; PMID:17205196; http://dx.doi.org/ 10.1100/tsw.2006.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, Heisenberg M, Fischbach KF, Pongs O. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 1995; 14(2):287-301; PMID:7857639; http://dx.doi.org/ 10.1016/0896-6273(95)90286-4 [DOI] [PubMed] [Google Scholar]
- 22. Degoutin JL, Milton CC, Yu E, Tipping M, Bosveld F, Yang L, Bellaiche Y, Veraksa A, Harvey KF. Riquiqui and minibrain are regulators of the hippo pathway downstream of Dachsous. Nat Cell Biol 2013; 15(10):1176-85; PMID:23955303; http://dx.doi.org/ 10.1038/ncb2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moller RS, Kubart S, Hoeltzenbein M, Heye B, Vogel I, Hansen CP, Menzel C, Ullmann R, Tommerup N, Ropers HH, et al. . Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am J Hum Genet 2008; 82(5):1165-70; PMID:18405873; http://dx.doi.org/ 10.1016/j.ajhg.2008.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hammerle B, Vera-Samper E, Speicher S, Arencibia R, Martinez S, Tejedor FJ. Mnb/Dyrk1A is transiently expressed and asymmetrically segregated in neural progenitor cells at the transition to neurogenic divisions. Dev Biol 2002; 246(2):259-73; PMID:12051815; http://dx.doi.org/ 10.1006/dbio.2002.0675 [DOI] [PubMed] [Google Scholar]
- 25. Hammerle B, Carnicero A, Elizalde C, Ceron J, Martinez S, Tejedor FJ. Expression patterns and subcellular localization of the Down syndrome candidate protein MNB/DYRK1A suggest a role in late neuronal differentiation. Eur J Neurosci 2003; 17(11):2277-86; PMID:12814361; http://dx.doi.org/ 10.1046/j.1460-9568.2003.02665.x [DOI] [PubMed] [Google Scholar]
- 26. Hammerle B, Elizalde C, Tejedor FJ. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur J Neurosci 2008; 27(5):1061-74; PMID:18364031; http://dx.doi.org/ 10.1111/j.1460-9568.2008.06092.x [DOI] [PubMed] [Google Scholar]
- 27. Yabut O, Domogauer J, D'Arcangelo G. Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J Neurosci 2010; 30(11):4004-14; PMID:20237271; http://dx.doi.org/ 10.1523/JNEUROSCI.4711-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soppa U, Schumacher J, Florencio Ortiz V, Pasqualon T, Tejedor FJ, Becker W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014; 13(13):2084-100; PMID:24806449; http://dx.doi.org/ 10.4161/cc.29104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hammerle B, Ulin E, Guimera J, Becker W, Guillemot F, Tejedor FJ. Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development 2011; 138(12):2543-54; PMID:21610031; http://dx.doi.org/ 10.1242/dev.066167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park J, Oh Y, Yoo L, Jung MS, Song WJ, Lee SH, Seo H, Chung KC. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J Biol Chem 2010; 285(41):31895-906; PMID:20696760; http://dx.doi.org/ 10.1074/jbc.M110.147520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat Rev Cancer 2007; 7(3):182-91; PMID:17318211; http://dx.doi.org/ 10.1038/nrc2070 [DOI] [PubMed] [Google Scholar]
- 32. Badouel C, Garg A, McNeill H. Herding Hippos: regulating growth in flies and man. Curr Opin Cell Biol 2009; 21(6):837-43; PMID:19846288; http://dx.doi.org/ 10.1016/j.ceb.2009.09.010 [DOI] [PubMed] [Google Scholar]
- 33. Reddy BV, Irvine KD. The Fat and Warts signaling pathways: new insights into their regulation, mechanism and conservation. Development 2008; 135(17):2827-38; PMID:18697904; http://dx.doi.org/ 10.1242/dev.020974 [DOI] [PubMed] [Google Scholar]
- 34. Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development 2011; 138(1):9-22; PMID:21138973; http://dx.doi.org/ 10.1242/dev.045500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu M, Zheng L, Han B, Wang L, Wang P, Liu H, Sun X. REST regulates DYRK1A transcription in a negative feedback loop. J Biol Chem 2011; 286(12):10755-63; PMID:21252229; http://dx.doi.org/ 10.1074/jbc.M110.174540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen JY, Lin JR, Tsai FC, Meyer T. Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol Cell 2013; 52(1):87-100; PMID:24119401; http://dx.doi.org/ 10.1016/j.molcel.2013.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang EJ, Ahn YS, Chung KC. Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J Biol Chem 2001; 276(43):39819-24; PMID:11518709; http://dx.doi.org/ 10.1074/jbc.M104091200 [DOI] [PubMed] [Google Scholar]
- 38. Kelly PA, Rahmani Z. DYRK1A enhances the mitogen-activated protein kinase cascade in PC12 cells by forming a complex with Ras, B-Raf, and MEK1. Mol Biol Cell 2005; 16(8):3562-73; PMID:15917294; http://dx.doi.org/ 10.1091/mbc.E04-12-1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fernandez-Martinez J, Vela EM, Tora-Ponsioen M, Ocana OH, Nieto MA, Galceran J. Attenuation of Notch signalling by the Down-syndrome-associated kinase DYRK1A. J Cell Sci 2009; 122(Pt 10):1574-83; PMID:19383720; http://dx.doi.org/ 10.1242/jcs.044354 [DOI] [PubMed] [Google Scholar]
- 40. Negrini S, Prada I, D'Alessandro R, Meldolesi J. REST: an oncogene or a tumor suppressor? Trends Cell Biol 2013; 23(6):289-95; PMID:23414932; http://dx.doi.org/ 10.1016/j.tcb.2013.01.006 [DOI] [PubMed] [Google Scholar]
- 41. Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005; 121(4):645-57; PMID:15907476; http://dx.doi.org/ 10.1016/j.cell.2005.03.013 [DOI] [PubMed] [Google Scholar]
- 42. Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, Lasorella A, Iavarone A, Chang S, Hernando E, et al. . Control of chromosome stability by the beta-TrCP-REST-Mad2 axis. Nature 2008; 452(7185):365-9; PMID:18354482; http://dx.doi.org/ 10.1038/nature06641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bahn S, Mimmack M, Ryan M, Caldwell MA, Jauniaux E, Starkey M, Svendsen CN, Emson P. Neuronal target genes of the neuron-restrictive silencer factor in neurospheres derived from fetuses with Down's syndrome: a gene expression study. Lancet 2002; 359(9303):310-5; PMID:11830198; http://dx.doi.org/ 10.1016/S0140-6736(02)07497-4 [DOI] [PubMed] [Google Scholar]
- 44. Canzonetta C, Mulligan C, Deutsch S, Ruf S, O'Doherty A, Lyle R, Borel C, Lin-Marq N, Delom F, Groet J, et al. . DYRK1A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic stem cell fate in Down syndrome. Am J Hum Genet 2008; 83(3):388-400; PMID:18771760; http://dx.doi.org/ 10.1016/j.ajhg.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lepagnol-Bestel AM, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N, Imbeaud S, Loe-Mie Y, Benihoud K, Agier N, et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum Mol Genet 2009; 18(8):1405-14; PMID:19218269; http://dx.doi.org/ 10.1093/hmg/ddp047 [DOI] [PubMed] [Google Scholar]
- 46. Ferron SR, Pozo N, Laguna A, Aranda S, Porlan E, Moreno M, Fillat C, de la Luna S, Sánchez P, Arbonés ML, et al. Regulated segregation of kinase Dyrk1A during asymmetric neural stem cell division is critical for EGFR-mediated biased signaling. Cell Stem Cell 2010; 7(3):367-79; PMID:20804972; http://dx.doi.org/ 10.1016/j.stem.2010.06.021 [DOI] [PubMed] [Google Scholar]
- 47. Aranda S, Alvarez M, Turro S, Laguna A, de la LS. Sprouty2-mediated inhibition of fibroblast growth factor signaling is modulated by the protein kinase DYRK1A. Mol Cell Biol 2008; 28(19):5899-911; PMID:18678649; http://dx.doi.org/ 10.1128/MCB.00394-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis 2008; 11(1):53-62; PMID:18219583; http://dx.doi.org/ 10.1007/s10456-008-9089-1 [DOI] [PubMed] [Google Scholar]
- 49. Rubin C, Zwang Y, Vaisman N, Ron D, Yarden Y. Phosphorylation of carboxyl-terminal tyrosines modulates the specificity of Sprouty-2 inhibition of different signaling pathways. J Biol Chem 2005; 280(10):9735-44; PMID:15637081; http://dx.doi.org/ 10.1074/jbc.M408308200 [DOI] [PubMed] [Google Scholar]
- 50. Abekhoukh S, Planque C, Ripoll C, Urbaniak P, Paul JL, Delabar JM, Janel N. Dyrk1A, a serine/threonine kinase, is involved in ERK and Akt activation in the brain of hyperhomocysteinemic mice. Mol Neurobiol 2013; 47(1):105-16; PMID:22923366; http://dx.doi.org/ 10.1007/s12035-012-8326-1 [DOI] [PubMed] [Google Scholar]
- 51. Guedj F, Pereira PL, Najas S, Barallobre MJ, Chabert C, Souchet B, Sebrie C, Verney C, Herault Y, Arbones M, et al. DYRK1A: a master regulatory protein controlling brain growth. Neurobiol Dis 2012; 46(1):190-203 PMID:22293606; http://dx.doi.org/ 10.1016/j.nbd.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 52. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81(3):323-30; PMID:7736585; http://dx.doi.org/ 10.1016/0092-8674(95)90385-2 [DOI] [PubMed] [Google Scholar]
- 53. Cobrinik D, Whyte P, Peeper DS, Jacks T, Weinberg RA. Cell cycle-specific association of E2F with the p130 E1A-binding protein. Genes Dev 1993; 7(12A):2392-404; PMID:8253385; http://dx.doi.org/ 10.1101/gad.7.12a.2392 [DOI] [PubMed] [Google Scholar]
- 54. Vairo G, Livingston DM, Ginsberg D. Functional interaction between E2F-4 and p130: evidence for distinct mechanisms underlying growth suppression by different retinoblastoma protein family members. Genes Dev 1995; 9(7):869-81; PMID:7705662; http://dx.doi.org/ 10.1101/gad.9.7.869 [DOI] [PubMed] [Google Scholar]
- 55. Smith EJ, Leone G, DeGregori J, Jakoi L, Nevins JR. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol Cell Biol 1996; 16(12):6965-76; PMID:8943352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, DeCaprio JA. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell 2007; 26(4):539-51; PMID:17531812; http://dx.doi.org/ 10.1016/j.molcel.2007.04.015 [DOI] [PubMed] [Google Scholar]
- 57. Sadasivam S, Decaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 2013; 13(8):585-95; PMID:23842645; http://dx.doi.org/ 10.1038/nrc3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Litovchick L, Florens LA, Swanson SK, Washburn MP, Decaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 2011; 25(8):801-13; PMID:21498570; http://dx.doi.org/ 10.1101/gad.2034211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tschop K, Conery AR, Litovchick L, Decaprio JA, Settleman J, Harlow E, Dyson N. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev 2011; 25(8):814-30; PMID:21498571; http://dx.doi.org/ 10.1101/gad.2000211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boichuk S, Parry JA, Makielski KR, Litovchick L, Baron JL, Zewe JP, Wozniak A, Mehalek KR, Korzeniewski N, Seneviratne DS, et al. . The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res 2013; 73(16):5120-9; PMID:23786773; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-0579 [DOI] [PubMed] [Google Scholar]
- 61. Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 2002; 32(1):148-52; PMID:12172547; http://dx.doi.org/ 10.1038/ng955 [DOI] [PubMed] [Google Scholar]
- 62. Hitzler JK, Cheung J, Li Y, Scherer SW, Zipursky A. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 2003; 101(11):4301-4; PMID:12586620; http://dx.doi.org/ 10.1182/blood-2003-01-0013 [DOI] [PubMed] [Google Scholar]
- 63. Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, Joazeiro PP, Saad ST, Costa FF. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet 2006; 38(7):807-12; PMID:16783379; http://dx.doi.org/ 10.1038/ng1825 [DOI] [PubMed] [Google Scholar]
- 64. Taub JW, Mundschau G, Ge Y, Poulik JM, Qureshi F, Jensen T, James SJ, Matherly LH, Wechsler J, Crispino JD. Prenatal origin of GATA1 mutations may be an initiating step in the development of megakaryocytic leukemia in Down syndrome. Blood 2004; 104(5):1588-9; PMID:15317736; http://dx.doi.org/ 10.1182/blood-2004-04-1563 [DOI] [PubMed] [Google Scholar]
- 65. Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, Crispino JD. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest 2012; 122(3):948-62; PMID:22354171; http://dx.doi.org/ 10.1172/JCI60455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, Orkin SH, Li Z. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev 2010; 24(15):1659-72; PMID:20679399; http://dx.doi.org/ 10.1101/gad.1903410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Maroz A, Stachorski L, Emmrich S, Reinhardt K, Xu J, Shao Z, Käbler S, Dertmann T, Hitzler J, Roberts I, et al. . GATA1s induces hyperproliferation of eosinophil precursors in Down syndrome transient leukemia. Leukemia 2014; 28(6):1259-70; PMID:24336126; http://dx.doi.org/ 10.1038/leu.2013.373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maenz B, Hekerman P, Vela EM, Galceran J, Becker W. Characterization of the human DYRK1A promoter and its regulation by the transcription factor E2F1. BMC Mol Biol 2008; 9:30; PMID:18366763; http://dx.doi.org/ 10.1186/1471-2199-9-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hasle H, Clemmensen IH, Mikkelsen M. Incidence of cancer in individuals with Down syndrome. Tidsskr Nor Laegeforen 2000; 120(24):2878-81; PMID:11143409 [PubMed] [Google Scholar]
- 70. Nizetic D, Groet J. Tumorigenesis in Down's syndrome: big lessons from a small chromosome. Nat Rev Cancer 2012; 12(10):721-32; PMID:22996602; http://dx.doi.org/ 10.1038/nrc3355 [DOI] [PubMed] [Google Scholar]
- 71. Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, et al. . NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006; 441(7093):595-600; PMID:16554754; http://dx.doi.org/ 10.1038/nature04678 [DOI] [PubMed] [Google Scholar]
- 72. Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park IH, Yoon SS, Minami T, et al. . Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 2009; 459(7250):1126-30; PMID:19458618; http://dx.doi.org/ 10.1038/nature08062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mancini M, Toker A. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer 2009; 9(11):810-20; PMID:19851316; http://dx.doi.org/ 10.1038/nrc2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a putative regulator of early T cell activation genes. Science 1988; 241(4862):202-5; PMID:3260404; http://dx.doi.org/ 10.1126/science.3260404 [DOI] [PubMed] [Google Scholar]
- 75. Muller MR, Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol 2010; 10(9):645-56; PMID:20725108; http://dx.doi.org/ 10.1038/nri2818 [DOI] [PubMed] [Google Scholar]
- 76. Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, Okamura H, Bolton D, Feske S, Hogan PG, et al. . A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 2006; 441(7093):646-50; PMID:16511445; http://dx.doi.org/ 10.1038/nature04631 [DOI] [PubMed] [Google Scholar]
- 77. Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 1997; 275(5308):1930-4; PMID:9072970; http://dx.doi.org/ 10.1126/science.275.5308.1930 [DOI] [PubMed] [Google Scholar]
- 78. Zhu J, Shibasaki F, Price R, Guillemot JC, Yano T, Dotsch V, Wagner G, Ferrara P, McKeon F. Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell 1998; 93(5):851-61; PMID:9630228; http://dx.doi.org/ 10.1016/S0092-8674(00)81445-2 [DOI] [PubMed] [Google Scholar]
- 79. Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A. A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Mol Cell Biol 2004; 24(10):4184-95; PMID:15121840; http://dx.doi.org/ 10.1128/MCB.24.10.4184-4195.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Birger Y, Izraeli S. DYRK1A in Down syndrome: an oncogene or tumor suppressor? J Clin Invest 2012; 122(3):807-10; PMID:22354166; http://dx.doi.org/ 10.1172/JCI62372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood 2014; 123(16):2451-9; PMID:24608975; http://dx.doi.org/ 10.1182/blood-2013-08-355818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lawinger P, Venugopal R, Guo ZS, Immaneni A, Sengupta D, Lu W, Rastelli L, Marin Dias Carneiro A, Levin V, Fuller GN, et al. . The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells. Nat Med 2000; 6(7):826-31; PMID:10888935; http://dx.doi.org/ 10.1038/77565 [DOI] [PubMed] [Google Scholar]
- 83. Su X, Gopalakrishnan V, Stearns D, Aldape K, Lang FF, Fuller G, Snyder E, Eberhart CG, Majumder S. Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol Cell Biol 2006; 26(5):1666-78; PMID:16478988; http://dx.doi.org/ 10.1128/MCB.26.5.1666-1678.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Palm K, Metsis M, Timmusk T. Neuron-specific splicing of zinc finger transcription factor REST/NRSF/XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Brain Res Mol Brain Res 1999; 72(1):30-9; PMID:10521596; http://dx.doi.org/ 10.1016/S0169-328X(99)00196-5 [DOI] [PubMed] [Google Scholar]
- 85. Conti L, Crisafulli L, Caldera V, Tortoreto M, Brilli E, Conforti P, Zunino F, Magrassi L, Schiffer D, Cattaneo E. REST controls self-renewal and tumorigenic competence of human glioblastoma cells. PLoS One 2012; 7(6):e38486; PMID:22701651; http://dx.doi.org/ 10.1371/journal.pone.0038486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kamal MM, Sathyan P, Singh SK, Zinn PO, Marisetty AL, Liang S, Gumin J, El-Mesallamy HO, Suki D, Colman H, et al. . REST Regulates Oncogenic Properties of Glioblastoma Stem Cells. Stem Cells 2012; 30(3):405-14; PMID:22228704; http://dx.doi.org/ 10.1002/stem.1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, Tan BK, Sethi G, Bishayee A. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta 2014; 1845(2):136-54; PMID:24388873; http://dx.doi.org/ 10.1016/j.bbcan.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 88. Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int 2013; 2013:421821; PMID:24199193; http://dx.doi.org/ 10.1155/2013/421821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wen Z, Zhong Z, Darnell JE, Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82(2):241-50; PMID:7543024; http://dx.doi.org/ 10.1016/0092-8674(95)90311-9 [DOI] [PubMed] [Google Scholar]
- 90. Matsuo R, Ochiai W, Nakashima K, Taga T. A new expression cloning strategy for isolation of substrate-specific kinases by using phosphorylation site-specific antibody. J Immunol Methods 2001; 247(1-2):141-51; PMID:11150545; http://dx.doi.org/ 10.1016/S0022-1759(00)00313-6 [DOI] [PubMed] [Google Scholar]
- 91. Sakaguchi M, Oka M, Iwasaki T, Fukami Y, Nishigori C. Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. J Invest Dermatol 2012; 132(7):1877-85; PMID:22418867; http://dx.doi.org/ 10.1038/jid.2012.45 [DOI] [PubMed] [Google Scholar]
- 92. Mao J, Maye P, Kogerman P, Tejedor FJ, Toftgard R, Xie W, Wu G, Wu D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J Biol Chem 2002; 277(38):35156-61; PMID:12138125; http://dx.doi.org/ 10.1074/jbc.M206743200 [DOI] [PubMed] [Google Scholar]
- 93. Altaba A. Hedgehog signaling and the Gli code in stem cells, cancer, and metastases. Sci Signal 2011; 4(200):t9; PMID:22114144; http://dx.doi.org/ 10.1126/scisignal.2002540 [DOI] [PubMed] [Google Scholar]
- 94. Seifert A, Allan LA, Clarke PR. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J 2008; 275(24):6268-80; PMID:19016842; http://dx.doi.org/ 10.1111/j.1742-4658.2008.06751.x [DOI] [PubMed] [Google Scholar]
- 95. Laguna A, Aranda S, Barallobre MJ, Barhoum R, Fernandez E, Fotaki V, Delabar JM, de la Luna S, de la Villa P, Arbonés ML. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev Cell 2008; 15(6):841-53; PMID:19081073; http://dx.doi.org/ 10.1016/j.devcel.2008.10.014 [DOI] [PubMed] [Google Scholar]
- 96. Pop C, Timmer J, Sperandio S, Salvesen GS. The apoptosome activates caspase-9 by dimerization. Mol Cell 2006; 22(2):269-75; PMID:16630894; http://dx.doi.org/ 10.1016/j.molcel.2006.03.009 [DOI] [PubMed] [Google Scholar]
- 97. Seifert A, Clarke PR. p38alpha- and DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site in response to hyperosmotic stress. Cell Signal 2009; 21(11):1626-33; PMID:19586613; http://dx.doi.org/ 10.1016/j.cellsig.2009.06.009 [DOI] [PubMed] [Google Scholar]
- 98. Imai SI, Guarente L. NAD and sirtuins in aging and disease. Trends Cell Biol 2014; 24(8):464-71; PMID:24786309; http://dx.doi.org/ 10.1016/j.tcb.2014.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem 2010; 285(17):13223-32; PMID:20167603; http://dx.doi.org/ 10.1074/jbc.M110.102574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ionescu A, Dufrasne F, Gelbcke M, Jabin I, Kiss R, Lamoral-Theys D. DYRK1A kinase inhibitors with emphasis on cancer. Mini Rev Med Chem 2012; 12(13):1315-29; PMID:23016545 [DOI] [PubMed] [Google Scholar]
- 101. Pozo N, Zahonero C, Fernandez P, Linares JM, Ayuso A, Hagiwara M, Pérez A, Ricoy JR, Hernández-Laín A, Sepúlveda JM, et al. . Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J Clin Invest 2013; 123(6):2475-87; PMID:23635774; http://dx.doi.org/ 10.1172/JCI63623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zahonero C, Sanchez-Gomez P. EGFR-dependent mechanisms in glioblastoma: towards a better therapeutic strategy. Cell Mol Life Sci 2014; PMID:24671641; http://dx.doi.org/ 10.1007/s00018-014-1608-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kales SC, Ryan PE, Nau MM, Lipkowitz S. Cbl and human myeloid neoplasms: the Cbl oncogene comes of age. Cancer Res 2010; 70(12):4789-94; PMID:20501843; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Barbachano A, Ordonez-Moran P, Garcia JM, Sanchez A, Pereira F, Larriba MJ, Martínez N, Hernández J, Landolfi S, Bonilla F, et al. . SPROUTY-2 and E-cadherin regulate reciprocally and dictate colon cancer cell tumourigenicity. Oncogene 2010; 29(34):4800-13; PMID:20543868; http://dx.doi.org/ 10.1038/onc.2010.225 [DOI] [PubMed] [Google Scholar]
- 105. Smith B, Medda F, Gokhale V, Dunckley T, Hulme C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: a new avenue for a disease modifying treatment of Alzheimer's? ACS Chem Neurosci 2012; 3(11):857-72; PMID:23173067; http://dx.doi.org/ 10.1021/cn300094k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hagiwara M. Alternative splicing: a new drug target of the post-genome era. Biochim Biophys Acta 2005; 1754(1-2):324-31; PMID:16260193; http://dx.doi.org/ 10.1016/j.bbapap.2005.09.010 [DOI] [PubMed] [Google Scholar]
- 107. Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007; 408(3):297-315; PMID:17850214; http://dx.doi.org/ 10.1042/BJ20070797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schenberg EE. Ayahuasca and cancer treatment. SAGE Open Med 2013; 1; http://dx.doi.org/ 10.1177/2050312113508389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hamsa TP, Kuttan G. Harmine inhibits tumour specific neo-vessel formation by regulating VEGF, MMP, TIMP and pro-inflammatory mediators both in vivo and in vitro. Eur J Pharmacol 2010; 649(1-3):64-73; PMID:20858484; http://dx.doi.org/10.1016/ [DOI] [PubMed] [Google Scholar]
- 110. Zaker F, Oody A, Arjmand A. A study on the antitumoral and differentiation effects of peganum harmala derivatives in combination with ATRA on leukaemic cells. Arch Pharm Res 2007; 30(7):844-9; PMID:17703736; http://dx.doi.org/ 10.1007/BF02978835 [DOI] [PubMed] [Google Scholar]
- 111. Dai F, Chen Y, Song Y, Huang L, Zhai D, Dong Y, Lai L, Zhang T, Li D, Pang X, et al. . A natural small molecule harmine inhibits angiogenesis and suppresses tumour growth through activation of p53 in endothelial cells. PLoS One 2012; 7(12):e52162; PMID:23300602; http://dx.doi.org/ 10.1371/journal.pone.0052162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Eid SY, El Readi MZ, Eldin EE, Fatani SH, Wink M. Influence of combinations of digitonin with selected phenolics, terpenoids, and alkaloids on the expression and activity of P-glycoprotein in leukaemia and colon cancer cells. Phytomedicine 2013; 21(1):47-61; PMID:23999162; http://dx.doi.org/ 10.1016/j.phymed.2013.07.019 [DOI] [PubMed] [Google Scholar]
- 113. Bei YY, Yuan ZQ, Zhang L, Zhou XF, Chen WL, Xia P, Liu Y, You BG, Hu XJ, Zhu QL, et al. . Novel self-assembled micelles based on palmitoyl-trimethyl-chitosan for efficient delivery of harmine to liver cancer. Expert Opin Drug Deliv 2014; 11(6):843-54; PMID:24655139; http://dx.doi.org/ 10.1517/17425247.2014.893292 [DOI] [PubMed] [Google Scholar]
- 114. Zhang H, Sun K, Ding J, Xu H, Zhu L, Zhang K, Li X, Sun W, et al. . Harmine induces apoptosis and inhibits tumor cell proliferation, migration and invasion through down-regulation of cyclooxygenase-2 expression in gastric cancer. Phytomedicine 2014; 21(3):348-55; PMID:24176842; http://dx.doi.org/ 10.1016/j.phymed.2013.09.007 [DOI] [PubMed] [Google Scholar]
- 115. Kim H, Sablin SO, Ramsay RR. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch Biochem Biophys 1997; 337(1):137-42; PMID:8990278; http://dx.doi.org/ 10.1006/abbi.1996.9771 [DOI] [PubMed] [Google Scholar]
- 116. Airaksinen MM, Lecklin A, Saano V, Tuomisto L, Gynther J. Tremorigenic effect and inhibition of tryptamine and serotonin receptor binding by beta-carbolines. Pharmacol Toxicol 1987; 60(1):5-8; PMID:3562389; http://dx.doi.org/ 10.1111/j.1600-0773.1987.tb01711.x [DOI] [PubMed] [Google Scholar]
- 117. Adayev T, Wegiel J, Hwang YW. Harmine is an ATP-competitive inhibitor for dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). Arch Biochem Biophys 2011; 507(2):212-8; PMID:21185805; http://dx.doi.org/ 10.1016/j.abb.2010.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ogawa Y, Nonaka Y, Goto T, Ohnishi E, Hiramatsu T, Kii I, Yoshida M, Ikura T, Onogi H, Shibuya H, et al. . Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat Commun 2010; 1:86; PMID:20981014; http://dx.doi.org/ 10.1038/ncomms1090 [DOI] [PubMed] [Google Scholar]
- 119. Gallego MA, Ballot C, Kluza J, Hajji N, Martoriati A, Castera L, Cuevas C, Formstecher P, Joseph B, Kroemer G, et al. . Overcoming chemoresistance of non-small cell lung carcinoma through restoration of an AIF-dependent apoptotic pathway. Oncogene 2008; 27(14):1981-92; PMID:17906690; http://dx.doi.org/ 10.1038/sj.onc.1210833 [DOI] [PubMed] [Google Scholar]
- 120. Baunbaek D, Trinkler N, Ferandin Y, Lozach O, Ploypradith P, Rucirawat S, Ishibashi F, Iwao M, Meijer L. Anticancer alkaloid lamellarins inhibit protein kinases. Mar Drugs 2008; 6(4):514-27; PMID:19172192; http://dx.doi.org/ 10.3390/md20080026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ballot C, Kluza J, Lancel S, Martoriati A, Hassoun SM, Mortier L, Vienne JC, Briand G, Formstecher P, Bailly C, et al. . Inhibition of mitochondrial respiration mediates apoptosis induced by the anti-tumoral alkaloid lamellarin D. Apoptosis 2010; 15(7):769-81; PMID:20151196; http://dx.doi.org/ 10.1007/s10495-010-0471-2 [DOI] [PubMed] [Google Scholar]
- 122. Echalier A, Bettayeb K, Ferandin Y, Lozach O, Clement M, Valette A, Liger F, Marquet B, Morris JC, Endicott JA, et al. Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J Med Chem 2008; 51(4):737-751; PMID:18232649; http://dx.doi.org/ 10.1021/jm700940h [DOI] [PubMed] [Google Scholar]
- 123. Jarry M, Lecointre C, Malleval C, Desrues L, Schouft MT, Lejoncour V, Liger F, Lyvinec G, Joseph B, Loaëc N, et al. . Impact of meriolins, a new class of cyclin-dependent kinase inhibitors, on malignant glioma proliferation and neo-angiogenesis. Neuro Oncol 2014; 16(11):1484-98 PMID:24891448; http://dx.doi.org/ 10.1093/neuonc/nou102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J 2003; 371(Pt 1):199-204; PMID:12534346; http://dx.doi.org/ 10.1042/BJ20021535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mercer SE, Friedman E. Mirk/Dyrk1B: a multifunctional dual-specificity kinase involved in growth arrest, differentiation, and cell survival. Cell Biochem Biophys 2006; 45(3):303-15; PMID:16845176; http://dx.doi.org/ 10.1385/CBB:45:3:303 [DOI] [PubMed] [Google Scholar]
- 126. Becker W. Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle 2012; 11(18):3389-94; PMID:22918246; http://dx.doi.org/ 10.4161/cc.21404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Friedman E. Mirk/Dyrk1B in cancer. J Cell Biochem 2007; 102(2):274-9; PMID:17583556; http://dx.doi.org/ 10.1002/jcb.21451 [DOI] [PubMed] [Google Scholar]
- 128. Gao J, Zheng Z, Rawal B, Schell MJ, Bepler G, Haura EB. Mirk/Dyrk1B, a novel therapeutic target, mediates cell survival in non-small cell lung cancer cells. Cancer Biol Ther 2009; 8(17):1671-9; PMID:19633423; http://dx.doi.org/ 10.4161/cbt.8.17.9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Friedman E. Mirk/dyrk1B Kinase in Ovarian Cancer. Int J Mol Sci 2013; 14(3):5560-75; PMID:23528858; http://dx.doi.org/ 10.3390/ijms14035560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res 2009; 69(8):3317-24; PMID:19351855; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Deng X, Mercer SE, Sun CY, Friedman E. The normal function of the cancer kinase Mirk/dyrk1B is to reduce reactive oxygen species. Genes Cancer 2014; 5(1-2):22-30; PMID:24955215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer 2009; 9(6):429-39; PMID:19472429; http://dx.doi.org/ 10.1038/nrc2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chen D, Wang CY, Lambert JD, Ai N, Welsh WJ, Yang CS. Inhibition of human liver catechol-O-methyltransferase by tea catechins and their metabolites: structure-activity relationship and molecular-modeling studies. Biochem Pharmacol 2005; 69(10):1523-31; PMID:15857617; http://dx.doi.org/ 10.1016/j.bcp.2005.01.024 [DOI] [PubMed] [Google Scholar]
- 134. Sanchez C, Salas AP, Brana AF, Palomino M, Pineda-Lucena A, Carbajo RJ, Méndez C, Moris F, Salas JA. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem Commun (Camb) 2009;(27):4118-20; PMID:19568652; http://dx.doi.org/ 10.1039/b905068j [DOI] [PubMed] [Google Scholar]
- 135. Neagoie C, Vedrenne E, Buron F, Merour JY, Rosca S, Bourg S, Lozach O, Meijer L, Baldeyrou B, Lansiaux A, et al. . Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues: a novel DYRK1A inhibitor class. Eur J Med Chem 2012; 49:379-96; PMID:22305342; http://dx.doi.org/ 10.1016/j.ejmech.2012.01.040 [DOI] [PubMed] [Google Scholar]
- 136. Akue-Gedu R, Debiton E, Ferandin Y, Meijer L, Prudhomme M, Anizon F, Moreau P. Synthesis and biological activities of aminopyrimidyl-indoles structurally related to meridianins. Bioorg Med Chem 2009; 17(13):4420-4; PMID:19477650; http://dx.doi.org/ 10.1016/j.bmc.2009.05.017 [DOI] [PubMed] [Google Scholar]
- 137. Giraud F, Alves G, Debiton E, Nauton L, Thery V, Durieu E, Ferandin Y, Lozach O, Meijer L, Anizon F, et al. . Synthesis, protein kinase inhibitory potencies, and in vitro antiproliferative activities of meridianin derivatives. J Med Chem 2011; 54(13):4474-89; PMID:21623630; http://dx.doi.org/ 10.1021/jm200464w [DOI] [PubMed] [Google Scholar]
- 138. Fedorov O, Huber K, Eisenreich A, Filippakopoulos P, King O, Bullock AN, Szklarczyk D, Jensen LJ, Fabbro D, Trappe J, et al. . Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem Biol 2011; 18(1):67-76; PMID:21276940; http://dx.doi.org/ 10.1016/j.chembiol.2010.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Debdab M, Renault S, Lozach O, Meijer L, Paquin L, Carreaux F, Bazureau JP. Synthesis and preliminary biological evaluation of new derivatives of the marine alkaloid leucettamine B as kinase inhibitors. Eur J Med Chem 2010; 45(2):805-10; PMID:19879673; http://dx.doi.org/ 10.1016/j.ejmech.2009.10.009 [DOI] [PubMed] [Google Scholar]
- 140. Fala F, Blalock WL, Tazzari PL, Cappellini A, Chiarini F, Martinelli G, Tafuri A, McCubrey JA, Cocco L, Martelli AM, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor (2S)-1-(1H-Indol-3-yl)-3-[5-(3-methyl-2H-indazol-5-yl)pyridin-3-yl]oxypropan2-ami ne (A443654) in T-cell acute lymphoblastic leukemia. Mol Pharmacol 2008; 74(3):884-95; PMID:18577685; http://dx.doi.org/ 10.1124/mol.108.047639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, et al. . The selectivity of inhibitors of protein kinase CK2: an update. Biochem J 2008; 415(3):353-65; PMID:18588507; http://dx.doi.org/ 10.1042/BJ20080309 [DOI] [PubMed] [Google Scholar]
- 142. Pagano MA, Andrzejewska M, Ruzzene M, Sarno S, Cesaro L, Bain J, Elliott M, Meggio F, Kazimierczuk Z, Pinna LA. Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J Med Chem 2004; 47(25):6239-47; PMID:15566294; http://dx.doi.org/ 10.1021/jm049854a [DOI] [PubMed] [Google Scholar]
- 143. Sarno S, de Moliner E, Ruzzene M, Pagano MA, Battistutta R, Bain J, Fabbro D, Schoepfer J, Elliott M, Furet P, et al. Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl]acetic acid (IQA). Biochem J 2003; 374(Pt 3):639-46; PMID:12816539; http://dx.doi.org/ 10.1042/BJ20030674 [DOI] [PMC free article] [PubMed] [Google Scholar]