

Abstract
Invalidation of uncoupling protein 2 (Ucp2) increases glucose utilization and proliferation in normal cells. We recently reported that cancer cells that overexpress UCP2 become less tumorigenic while switching their metabolism from glycolysis to oxidative phosphorylation. UCP2 appears to be a key regulator of cellular metabolism with a relevant function against tumorigenesis.
Keywords: uncoupling protein 2, mitochondria, cancer, proliferation, metabolism reprogramming
Metabolic remodeling associated with cancer is the subject of renewed research interest that integrates multiple aspects of bioenergetic adaptation. The maintenance of mitochondrial function during cell growth and development is tightly associated with mitochondria–nuclear crosstalk. Because of the quantitatively predominant role of the nuclear genome in mitochondria biogenesis, much attention over the past several decades has been directed to the analysis of anterograde regulation. However, recent studies have revealed that mitochondria are also engaged in retrograde regulation, which can be defined as cellular responses, mostly changes in nuclear gene expression, to changes in the functional state of mitochondria. This retrograde signaling is for the most part an adaptive response and its outcome is usually a recasting of metabolic, regulatory, or stress-related pathways.
Uncoupling protein 2 (UCP2) is a mitochondrial carrier whose protein expression is tightly related to changes in cell proliferation, and as such is a crucial player in the cascade of mitochondrial molecular events associated with carcinogenesis. Indeed, Ucp2 invalidation is associated with increased cell proliferation both in primary embryonic fibroblasts (MEF) and in activated T cells isolated from Ucp2−/− mice.1 In our recent report in Cancer Research2 we showed that direct manipulation of mitochondrial activity through expression of this inner membrane carrier induces a feed-forward loop from mitochondria to the adenosine monophosphate-activated protein kinase (AMPK)/hypoxia inducible factor (HIF) axis that modifies cancer cell proliferation (Fig. 1). Using different cancer cell lines that overexpress UCP2, we showed that UCP2 protein expression level correlates closely and negatively with tumor proliferation in vitro and in vivo. This decrease in proliferation is associated with metabolic remodeling, i.e., decreased glycolysis and increased oxidative phosphorylation. We showed that the antitumor effect of UCP2 is associated with an increase in AMPK signaling and a decrease in HIF. Our study indicated that a reduced fumarate level driven by UCP2 could be the link between AMPK activation and the significant decrease in HIF2-α expression. Indeed, cancers carrying mutations in enzymes involved in the tricarboxylic acid cycle (TCA), specifically succinate dehydrogenase (SDH) and fumarate hydratase (FH), show intracytoplasmic accumulation of fumarate and succinate that inhibits the prolyl hydroxylase domain proteins (PHD), thus allowing stabilization of HIF.3 Furthermore, other studies have shown that AMPK activity is decreased in renal cancer cells deficient in FH enzyme, which also accumulate fumarate.4
Figure 1.
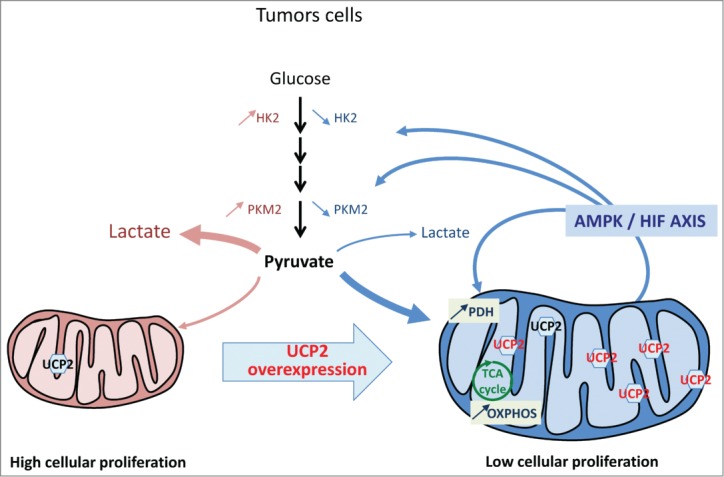
UCP2-induced metabolic reprogramming involves the HIF/AMPK axis to inhibit proliferation of cancer cells. Tumor cells with low protein levels of endogenous uncoupling protein 2 (UCP2) proliferate rapidly and express high levels of hexokinase 2 (HK2) and pyruvate kinase isoform 2 (PKM2) enzymes. In these cells, UCP2 overexpression triggers a metabolic reprogramming favoring oxidative metabolism with increased expression of pyruvate dehydrogenase (PDH) and oxidative phosphorylation (OXPHOS), and conversely decreased expression of HK2 and PKM2. This reprogramming is associated with decreased hypoxia inducible factor (HIF) signaling and increased adenosine monophosphate-activated protein kinase (AMPK) activity. This feed-forward loop from mitochondria to AMPK/HIF axis driven by UCP2 decreases the tumorigenic properties of tumor cells.
Despite the answers provided by our investigation, several questions remain: Which metabolite does UCP2 transport? How is AMPK activated? Does such UCP2 retrograde signaling apply to all tumors? Could a UCP2 activator have beneficial effects on cancer treatment, either by slowing down cancer progression or by sensitizing cancer cells to treatment?
UCP2 activity has long been associated with an uncoupling effect.5 In our study, UCP2 overexpression does not alter mitochondrial membrane potential, or the P/O ratio between ATP synthesis and oxygen consumption. Instead, UCP2 modifies metabolic substrate orientation through modulation of metabolite compartmentalization involved in mitochondrial anaplerotic metabolism. Moreover, in support of this hypothesis, Fiermonte's laboratory recently showed that UCP2 protein transports 4-carbon metabolites such as aspartate, malate, and oxaloacetate out of the mitochondria.6
Our results suggest that re-expression or reactivation of UCP2 may be a winning therapeutic strategy. However, several studies have previously shown that UCP2 overexpression in cancer cells was associated with increased proliferation,7-9 decreased ROS production, and decreased apoptosis8,9 whereas other studies showed that UCP2 silencing reduced survival through increased differentiation and apoptosis.10 However, all of these studies were performed in cancer cell lines expressing a high endogenous level of UCP2, such as colon, breast, and lung cancer cells. In contrast, our study is the only one using cancer cells with a low endogenous UCP2 protein level and in these cells UCP2 overexpression has an antiproliferative effect. In fact, microarray database analysis (www.oncomine.org) allows the distinction of 2 different types of cancer on the basis of UCP2 mRNA expression: cancers with high UCP2 mRNA level such as lymphomas, lung, breast, and colorectal cancers; and cancers with low UCP2 mRNA level such as glioblastoma, melanoma, prostate, and liver cancers. This observation may underlie some metabolic differences between these 2 groups. However, mRNA expression does not always reflect UCP2 protein level since UCP2 is translationally regulated and it would therefore be interesting to determine whether UCP2 protein level is concordant with its mRNA expression in these cancers. Together, these observations suggest that UCP2 involvement in tumorigenesis is cell type-dependent and may depend on the metabolism exhibited by normal cells before their transformation, as reflected by the endogenous protein level of UCP2. In addition, it will be important to determine whether UCP2 protein expression is correlated with tumor grade, tumor invasiveness, and energy metabolism.
Finally, the development of animal models for different cancers, in particular those with low (e.g., glioblastoma) and high (e.g., colorectal) UCP2 expression, is essential in order to understand whether UCP2 is able to modify tumor metabolism in vivo and to determine more precisely which metabolite is transported by UCP2. Moreover, in vivo models will highlight whether (i) UCP2 function affects crucial cellular players in cancer; (ii) UCP2 could be considered as a biomarker to distinguish metabolism of tumors; and, most importantly, (iii) targeting UCP2 can improve therapeutic outcomes.
Our study contributes to the understanding of cancer metabolism. UCP2 appears to be an interesting candidate in the dialog between energy metabolism and proliferation. Moreover, since the mitochondrial activity of cells is directly associated with their susceptibility to different agents that induce cell death (i.e., chemotherapy or radiotherapy) UCP2 may be a novel target for sensitization of tumor cells to anticancer treatments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was financially supported by grants from the “Center National de la Recherche Scientifique” (CNRS), by the “Institut National de la Santé et de la Recherche Médicale” (Inserm), by the University Paris Descartes and by the Ministère de la Recherche.
References
- 1. Pecqueur C, Bui T, Gelly C, Hauchard J, Barbot C, Bouillaud F, Ricquier D, Miroux B, Thompson CB. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J 2008; 22:9-18; PMID:17855623; http://dx.doi.org/ 10.1096/fj.07-8945com [DOI] [PubMed] [Google Scholar]
- 2. Esteves P, Pecqueur C, Ransy C, Esnous C, Lenoir V, Bouillaud F, Bulteau AL, Lombes A, Prip-Buus C, Ricquier D. et al. . Mitochondrial retrograde signaling mediated by UCP2 inhibits cancer cell proliferation and tumorigenesis. Cancer Res 2014; 74:3971-82; PMID:24853548; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-3383 [DOI] [PubMed] [Google Scholar]
- 3. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 2006; 25:4675-82; PMID:16892081; http://dx.doi.org/ 10.1038/sj.onc.1209594 [DOI] [PubMed] [Google Scholar]
- 4. Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, Romero VV, Sougrat R, Vaulont S, Viollet B. et al. . The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 2011; 20:315-27; PMID:21907923; http://dx.doi.org/ 10.1016/j.ccr.2011.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB. et al. . Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 2001; 105:745-55; PMID:11440717; http://dx.doi.org/ 10.1016/S0092-8674(01)00378-6 [DOI] [PubMed] [Google Scholar]
- 6. Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D. et al. . UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci U S A 2014; 111:960-5.PMID:24395786; http://dx.doi.org/ 10.1073/pnas.1317400111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ayyasamy V, Owens KM, Desouki MM, Liang P, Bakin A, Thangaraj K, Buchsbaum DJ, LoBuglio AF, Singh KK. Cellular model of Warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PloS one 2011; 6:e24792; PMID:21935467; http://dx.doi.org/ 10.1371/journal.pone.0024792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deng S, Yang Y, Han Y, Li X, Wang X, Li X, Zhang Z, Wang Y. UCP2 inhibits ROS-mediated apoptosis in A549 under hypoxic conditions. PloS one 2012; 7:e30714; PMID:22292025; http://dx.doi.org/ 10.1371/journal.pone.0030714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Derdak Z, Mark NM, Beldi G, Robson SC, Wands JR, Baffy G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res 2008; 68:2813-9; PMID:18413749; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sayeed A, Meng Z, Luciani G, Chen LC, Bennington JL, Dairkee SH. Negative regulation of UCP2 by TGFbeta signaling characterizes low and intermediate-grade primary breast cancer. Cell Death Dis 2010; 1:e53; PMID:21364658; http://dx.doi.org/ 10.1038/cddis.2010.30 [DOI] [PMC free article] [PubMed] [Google Scholar]