

Abstract
Our recent study suggests that targeting GD3 synthase (also known as ST8SIA1)—the rate-limiting enzyme in biosynthesis of the breast cancer stem cell marker GD2—abrogates metastasis and depletes the cancer stem cell populations within a tumor, thus providing an effective therapeutic strategy against metastatic breast cancers.
Keywords: cancer stem cells, EMT, GD2, GD3S, metastasis
Abbreviations and acronyms
- CSC
cancer stem cell
- EMT
epithelial-mesenchymal transition
- FOXC2
forkhead box protein C2
- GalNAc
N-acetylgalactosamine
- GD2
ganglioside GD2 (disialoganglioside)
- GD3
ganglioside GD3 (disialoganglioside)
- GD2S
GD2 synthase
- GD3S
GD3 synthase
- GM3
ganglioside GM3 (monosialodihexosylganglioside)
- NF-KB
nuclear factor-κB
- shRNA
short hairpin ribonucleic acid
- TGFβ
transforming growth factor β
In the last decade or so, it has become increasingly accepted that tumor growth is sustained by a small subpopulation of tumor cells called cancer stem cells (CSCs) that are endowed with self-renewal and tumor-initiating capabilities.1 The inherent resilience of CSCs is believed to underpin tumor recurrence, therapy resistance, and metastasis with the corollary that the bulk of the tumor is composed of a mixture of proliferating and post-mitotic differentiated cells that have little consequence on tumor sustenance in the longer term. Acceptance of the CSC concept has re-directed therapeutic strategies toward the elusive CSC subset. In addition, it has become clear that targeting solely the tumor bulk may not only fail to eradicate disease but may also inadvertently enrich for therapy-resistant CSCs and lead to tumor recurrence.2
Although the CSC paradigm holds great promise for the development of novel and more effective therapies,1 the study of CSCs has been hampered not only by the lack of definitive markers but also by the fact that the CSC phenotype is intrinsically unstable, with tumor cells oscillating between distinct tumor-initiating/CSC and differentiated/non-CSC states.3
We recently showed that the aberrant activation of a latent embryonic program, known as the epithelial-mesenchymal transition (EMT), bestows stem cell properties and confers the migratory and invasive capabilities that underpin metastatic competence.4 In addition, we found that cells that have undergone EMT exhibit phenotypic and functional characteristics of mesenchymal stem cells5 and present the ganglioside GD2 on the cell surface.6 As such, we identified GD2 as a novel breast CSC marker that could be used not only for the isolation of CSCs, but also for their eradication. We demonstrated that inhibition of GD3 synthase (GD3S)—the rate-limiting enzyme for GD2 biosynthesis—using either short hairpin ribonucleic acid (shRNA) or a small molecule known as triptolide reduced the proportion of CSCs and abrogated primary tumor formation.6 However, the role of GD3S and GD2 in EMT and metastasis and, in particular, whether targeting GD3S could alleviate metastasis—the principal cause of cancer-related mortality—remained unclear.
A key finding of the present study by Sarkar et al.7 is that GD3S lies at the epicenter of multiple EMT signaling pathways since its inhibition prevents not only the initiation of multiple EMT programs but also the maintenance of the established mesenchymal phenotype. This suggests that targeting GD3S could serve to both inhibit the de novo generation of CSCs via EMT and to revert existing CSCs to a more differentiated, therapeutically vulnerable state.
At the molecular level, the transcription factor forkhead box protein C2 (FOXC2), a key downstream effector of several EMT pathways,8 transcriptionally regulates GD3S expression, directly linking inflammatory nuclear factor κB (NF-κB) signaling to activation of GD3S and the generation of GD2-positive CSCs. This is consistent with the purported roles of inflammation in exacerbating tumor progression. We also found that GD3S functions via c-Met signaling to promote EMT. Whereas GD3S levels correlate with c-Met activation in mesenchymal cells, GD3S inhibition elicits a significant decrease in levels of active phospho-c-Met and its downstream effector phospho-Akt. Although it remains unclear how engagement of GD2 signaling drives the phosphorylation/activation of c-Met, these findings link GD3S/GD2 with key players of a CSC-specific signaling pathway (Fig. 1).
Figure 1.
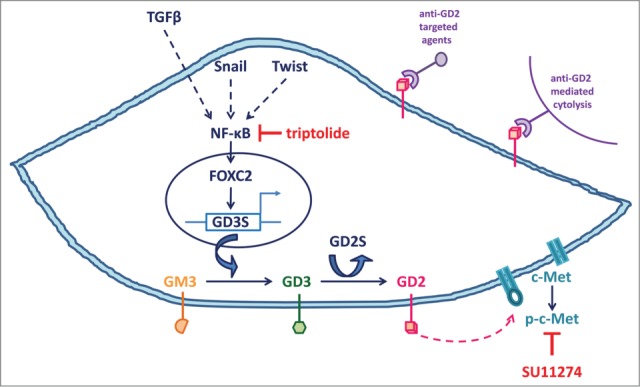
Key players in the GD3 synthase (GD3S)/ganglioside GD2 (GD2)-dependent signaling pathway that regulates stemness, the epithelial-mesenchymal transition (EMT), and metastatic dissemination in cancer cells. Extracellular EMT-inducing signals (e.g., transforming growth factor β [TGFβ]) and inflammation elicit the activation of EMT-inducing transcription factors (Snail, Twist) and nuclear factor KB (NF-KB) that coordinately promote the expression of the central EMT-mediator forkhead box protein C2 (FOXC2), which transcriptionally activates GD3S. GD3S is an enzyme that catalyzes the transfer of sialic acid from cytidine monophosphate-sialic acid to GM3 to produce the ganglioside GD3 (GD3), which is subsequently converted to GD2 via the activity of GD2 synthase (GD2S) and the transfer of N-acetylgalactosamine (GalNAc). Although GD2S is directly responsible for GD2 biosynthesis, GD3S is the rate-limiting enzyme for GD2 biosynthesis in cells that have undergone EMT and mesenchymal breast cancer cells. Targeting GD3S and/or c-Met signaling using triptolide or SU11274, respectively, reverts the combined EMT/cancer stem cell (CSC) phenotype and abrogates metastasis. Anti-GD2–based immunotherapies may also be used to eradicate CSCs. Broken arrows indicate unknown intermediate steps.
Histological analyses revealed that GD2 is localized at the tumor invasive front that is thought to foster cells actively undergoing EMT,9 and that GD3S inhibition markedly reduced invasion of the tumor into the surrounding tissue. Accordingly, inhibition of GD3S compromises several in vitro properties associated with metastatic competence including migration, invasion, and mammosphere formation, a surrogate assay for the attributes of stem and progenitor cells. Most importantly, genomic and pharmacological inhibition of GD3S compromised metastasis in both experimental and spontaneous metastasis models in the context of an intact immune system. Taken together, these results implicate GD3S in multiple facets of the intrinsic metastatic capabilities of tumor cells.
Although we observed a marked reduction in the incidence of metastasis following GD3S inhibition using shRNA or triptolide, we noted the presence of a small number of solitary disseminated tumor cells deposited in the lungs. These findings may reflect incomplete inhibition of GD3S in our tumor models. Nevertheless, these results have high clinical relevance since the expression of high levels of GD3S and phospho-c-Met correlate with poor prognosis in triple-negative human breast tumors, a particularly aggressive form of breast cancer with high rates of local and distant relapse.
Exploiting GD2 as a cell surface antigen of breast CSCs not only enables their isolation but also presents a tangible therapeutic opportunity, not least because the GD2 antigen is linked to CSC functionality. The development of high affinity anti-GD2 monoclonal antibodies is already underway, with emerging therapeutic strategies focusing on the use of these antibodies to stimulate an immune response against tumors by triggering monoclonal antibody-mediated cytolysis or to effect GD2-targeted delivery of therapeutic and diagnostic agents.10 While the effectiveness of immunotherapies based on anti-GD2 antibodies may be somewhat limited by the inability of the antibodies to penetrate solid tumors, our results raise the realistic possibility that small molecules that target GD3S and/or c-Met signaling may be used, in combination with standard-of-care therapies targeting the tumor bulk, to curb breast cancer recurrence and metastatic propensity.
Conflict of Interest
TRS, VLB, MA and SAM are inventors of a patent application based on the work described here. NS declares no conflict of interest.
Funding
Studies in SAM's laboratory are supported by NIH/NCI CA155243-01 and the American Cancer Society M. Patricia Alexander Research Scholar award (121958-RSG-12-102-01-DDC).
References
- 1.McDermott SP, Wicha MS. Targeting breast cancer stem cells. Mol Oncol 2010; 4:404-19; PMID:20599450; http://dx.doi.org/ 10.1016/j.molonc.2010.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A 2009; 106:13820-5; PMID:19666588; http://dx.doi.org/ 10.1073/pnas.0905718106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D'Alessio AC, Young RA, Weinberg RA. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013; 154:61-74; PMID:23827675; http://dx.doi.org/ 10.1016/j.cell.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704-15; PMID:18485877; http://dx.doi.org/ 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, Wang RY, Brisken C, Guerra R, Andreeff M, et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 2010; 28:1435-45; PMID:20572012; http://dx.doi.org/ 10.1002/stem.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, Guerra R, Sahin AA, Marini FC, Hortobagyi G, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest 2012; 122:2066-78; PMID:22585577; http://dx.doi.org/ 10.1172/JCI59735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar TR, Battula VL, Werden SJ, Vijay GV, Ramirez-Pena EQ, Taube JH, Chang JT, Miura N, Porter W, Sphyris N, et al. GD3 synthase regulates epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene 2014; 0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res 2013; 73:1981-92; PMID:23378344; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer 2005; 5:744-9; PMID:16148886; http://dx.doi.org/ 10.1038/nrc1694 [DOI] [PubMed] [Google Scholar]
- 10.Ahmed M, Cheung NK. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Lett 2014; 588:288-97; PMID:24295643; http://dx.doi.org/ 10.1016/j.febslet.2013.11.030 [DOI] [PubMed] [Google Scholar]