

Abstract
Non-oxidative glucose metabolism represents a hallmark of cancer. It is now apparent that different cell states among normal stem or progenitor cells have distinct aerobic glycolysis (AG) dependencies. However, malignant cells are markedly more vulnerable to modifications of AG regardless of the differentiation state of their cell of origin.
Keywords: metabolism, aerobic glycolysis, stem cells, progenitor cells, hematopoiesis, leukemia
How glucose is metabolized can influence cell function, but whether differences in glucose metabolism reflect, or dictate, the cell state is not clear and of particular interest given the association of cancer with aerobic glycolysis (AG). Cancer cells consume more glucose than non-proliferating cells but preferentially ferment glucose to lactate regardless of oxygen availability. This phenomenon was initially observed by Otto Warburg and now bears his name.1 Why cancer cells employ a less efficient metabolic pathway, at least for energy production, has been a longstanding puzzle although it is now believed that tumor cells consume glucose for purposes beyond ATP generation. Proliferating cells must convert nutrients, including glucose, into anabolic intermediates for macromolecule biosynthesis during the generation of new cells. Compared with mitochondrial respiration, AG drives this process through a more rapid metabolic flux that facilitates anabolism and may maintain a more favorable redox balance to serve robust cell proliferation.
The molecular basis for the Warburg effect in cancer cells has been attributed to a specific isoform of pyruvate kinase, PKM2.2 PK catalyzes conversion of phosphoenolpyruvate to pyruvate. In mammals, the M1 and M2 isoforms are alternatively spliced products of the PKM gene. Pyruvate kinase M1 (PKM1) is expressed in tissues with high ATP demand and promotes glucose metabolism via oxidative phosphorylation. PKM2 is expressed in embryonic tissues, cancers, and adult cells that exhibit high anabolic activity.3 Whereas PKM1 exists as a constitutively active tetramer, PKM2 can allosterically switch between a high-activity tetramer and a low-activity non-tetramer. In proliferating cells, PKM2 predominantly exists in non-tetrameric forms.4 Replacing PKM2 with PKM1 or stabilizing the PKM2 tetramer by small molecules reverses the Warburg effect and inhibits tumor cell growth.2,4
Normal somatic cells that are thought to also preferentially use glycolytic metabolism include tissue stem cells, particularly the self-renewing hematopoietic stem cells (HSC) resident in the hypoxic microenvironment of the bone marrow. We initially hypothesized that the metabolic demands of stem/progenitor cells were comparable to those of malignant cells, and therefore targeting metabolic pathways as proposed for solid tumors might result in disruption of normal tissue integrity. In other words, is the “Warburg effect” observed in cancer truly cancer cell-specific or is it exploiting a pathway that is important for normal stem/progenitor cell function?
To address this question, we conditionally deleted the Pkm2-specific exon 10 in mouse hematopoietic cells.5 Loss of Pkm2 led to expression of Pkm1 in all cells due to derepression of exon 9. The switch of PK isoforms was associated with a mild inhibition of AG and increased oxidative phosphorylation. Despite these metabolic changes, however, Pkm2 deletion did not have a detectable effect on normal hematopoiesis under homeostatic conditions. When tested under the stress of serial transplantation, Pkm2 depletion moderately affected the long-term bone marrow repopulation capacity due to impaired progenitor cell expansion. Interestingly, there was no effect on HSC maintenance. These results suggest that HSCs are less sensitive than progenitor cells to modulation of AG.
PKM2 has been reported to have non-metabolic functions in certain cancer cells. To confirm that inhibition of glycolysis indeed caused a progenitor defect, we engineered a mouse strain with knockout of lactate dehydrogenase A (Ldha), a more potent mediator of AG that functions downstream of Pkm2. In solid tumor models, inhibition of LDHA by RNAi or small molecules suppressed AG, caused oxidative stress, and blocked tumor progression.6 In mouse hematopoietic cells, Ldha deletion almost completely inhibited lactate production but did not cause changes in HSC number under homeostatic conditions.5 However, stress again revealed a dependency, this time in both progenitor populations and stem cells. The stem cell defect was associated with increased mitochondrial activity and generation of reactive oxygen species and could be partially rescued by antioxidant.
Further pushing the question of whether metabolic changes had different cell state dependencies, we decided to test how leukemia affecting each of the two cell states (progenitor and stem cells) would tolerate a change in AG. We tested two leukemia models, chronic myeloid leukemia (CML) and acute myeloid leukemia (AML), which are driven by BCR-ABL–transformed stem cells and MLL-AF9–transformed progenitor cells respectively.7,8 In both models, deletion of Pkm2 or Ldha markedly delayed leukemia initiation.5 Importantly, leukemia cells were far more sensitive than normal cells when tested in a direct competition experiment in vivo, suggesting that pharmacologically targeting either PKM2 or LDHA in leukemia may have a substantial impact on the malignant cells while sparing normal hematopoietic stem and progenitor cells.
The anticancer effect of Ldha depletion was also reported in a mouse lung cancer model.9 In this study, loss of Ldha impacted tumor-initiating cells. It remains to be determined, however, whether deletion of Ldha also impairs the function of normal lung epithelial cells. Interestingly, a different phenotype was observed in a breast cancer model driven by loss of function of Bcra1 (breast cancer 1, early onset), in which deletion of Pkm2 accelerated tumorigenesis.10 In contrast to leukemia, deletion of Pkm2 in breast cancer resulted in Pkm1 expression only in a subset of tumor cells that were negative for the proliferation marker proliferating cell nuclear antigen (PCNA).10 All PCNA-positive cells were negative for PKM, suggesting a selection against high PK activity in proliferating cells. It is not clear why some tumor cells in this model did not express Pkm1 in the absence of Pkm2. The results of both studies, however, support the notion that low PK activity may be required for cancer cells to proliferate.2,4 Pkm2 therefore serves as a tunable enzyme with the ability to adjust the level of AG to meet different cellular needs.
In summary, our results indicate differential dependencies on AG according to cell state among primary normal cells of defined differentiation status and between normal and malignant cells of a particular differentiation status (Fig. 1). These findings point to both important biological implications and potential therapeutic opportunities, as malignant cells revealed a definitive vulnerability not seen in their normal counterparts. A few questions are raised by this study: What are the splicing mechanisms that lead to Pkm2 expression in normal hematopoietic cells and leukemic cells and how are they regulated? Does deletion of Pkm2 or Ldha impair leukemia-initiating cells? How does inhibition of glycolysis compromise specific biosynthetic pathways in leukemic cells? Do the metabolic changes induced by loss of Pkm2 or Ldha affect other pathways regulating cell growth, survival, and energy sensing? Answering these questions will help us better understand the metabolic requirements of normal and malignant progenitor/stem cells and design novel strategies to treat leukemia with minimum toxicity.
Figure 1.
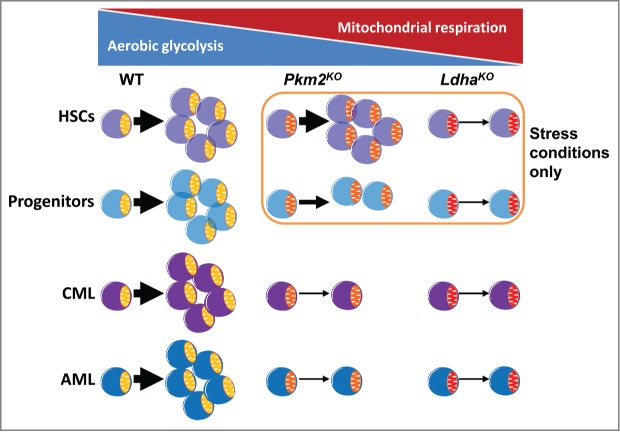
Cell state-specific dependency on aerobic glycolysis (AG). Deletion of pyruvate kinase M2 (Pkm2) moderately inhibits AG and mildly impairs progenitor cell expansion without affecting hematopoietic stem cells (HSCs). Knockout of lactate dehydrogenase A (Ldha) severely blocks AG and compromises both progenitor and stem cell function. The effects on normal stem and progenitor cells are only apparent under stress conditions. Leukemia is sensitive to even moderate inhibition of glycolysis regardless of cell origin. WT, wild type; Pkm2KO, Pkm2 knockout; LdhaKO, Ldha knockout; CML, chronic myeloid leukemia; AML, acute myeloid leukemia.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a NIH-HSCI T32 Training Grant (5T32HL87735-4) and an American Cancer Society postdoctoral fellowship (Y.H.W) and by NIH grants DK050234, HL044851 and CA148180 (D.T.S).
References
- 1.Warburg O. Origin of cancer cells. Oncologia 1956; 9:75-83; PMID:13335077; CrossRef [PubMed] [Google Scholar]
- 2.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008; 452:230-3; PMID:18337823; CrossRef [DOI] [PubMed] [Google Scholar]
- 3.Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A 2010; 107:1894-9; PMID:20133837; CrossRef [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 2012; 8:839-47; PMID:22922757; CrossRef [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang YH, Israelsen WJ, Lee D, Yu VW, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG, Scadden DT. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014; 158:1309-23; PMID:25215489; CrossRef [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010; 107:2037-42; PMID:20133848; CrossRef [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi N, Miura I, Saitoh K, Miura AB. Lineage involvement of stem cells bearing the philadelphia chromosome in chronic myeloid leukemia in the chronic phase as shown by a combination of fluorescence-activated cell sorting and fluorescence in situ hybridization. Blood 1998; 92:4758-63; PMID:9845542 [PubMed] [Google Scholar]
- 8.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006; 442:818-22; PMID:16862118; CrossRef [DOI] [PubMed] [Google Scholar]
- 9.Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, et al. Targeting lactate dehydrogenase–a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab 2014; 19:795-809; PMID:24726384; CrossRef [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW, et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013; 155:397-409; PMID:24120138; CrossRef [DOI] [PMC free article] [PubMed] [Google Scholar]