

Abstract
Cancer cells have an increased reliance on lipogenesis, which is required for uncontrolled cell division. We recently reported transcriptional and functional ‘reprogramming’ of the cellular energy grid, allowing cancer cells to divert metabolism from biosynthesis to bioenergetic pathways and thus supplying enhanced mobility during epithelial–mesenchymal transition (EMT) induced by transforming growth factor β (TGF-β1) (Fig. 1).
Keywords: Epithelial-to-Mesenchymal Transition, fatty acid synthase, lipogenesis, metastasis, transforming growth factor beta 1 (TGFβ1)
Unlike most differentiated non-proliferating cells, cancer cells tend to display a high glycolytic rate regardless of the presence or absence of oxygen, a phenomenon known as the ‘Warburg effect’. Cancer cells use glycolysis for energy and to generate building blocks for cell division.1 Lipids, amino acids, and nucleotides are the 3 basic elements required for proliferation. Lipogenesis is commonly achieved by increased de novo fatty acid synthesis to support rapid cell membrane growth.2
Although primary tumors can be removed by surgery, metastatic disease is the major cause of death for most cancers, and particularly breast and lung carcinomas. To date, most findings on cancer metabolism have been derived from primary tumors and cancer cell lines. Although metabolism has been studied in vivo in gliomas3, these tumors do not typically metastasize and therefore do not instruct about metabolic transitions that may accompany the acquisition of metastatic capacity. The major focus of our studies is stromal–tumor microenvironmental interactions that affect cancer metabolism. Using a transforming growth factor β1 (TGF-β1)-induced epithelial–mesenchymal transition (EMT) model, we found a metabolic profile in migrating cells that differs from that in rapidly proliferating cells.4 TGF-β1 exposure of A549 non-small cell lung cancer (NSCLC) cells, which induced an EMT, resulted in coordinated transcriptional suppression of lipogenic enzymes, including fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC). These enzymes are normally under the control of the lipogenic master transcriptional regulators carbohydrate-responsive element-binding protein (ChREBP) and sterol regulatory element binding proteins (SREBPs). We noted dramatic downregulation of both SREBPs and ChREBP during TGF-β1-induced EMT in A549 cells. SNAIL1, a well-known key transcription factor in the regulation of EMT,5 mediated suppression of this lipogenic reprogramming. In the absence of TGF-β1 treatment, SNAIL1 overexpression decreased lipogenesis by dramatically repressing ChREBP levels. These investigations are among the first to show the relationship between EMT and energy metabolism at a transcriptional level.
Simultaneously, Dr. Raghu Kalluri's group reported that the transcriptional co-activator peroxisome proliferator-activated receptor gamma, co-activator 1alpha (PGC-1α) promoted metastasis in an in vivo orthotopic implantation mouse model.6 They found that the lipogenic enzymes Fasn and Acc were expressed at very low levels in circulating cancer cells (CCCs), compared to their higher expression profile in primary cancer cells (PCCs). Although PGC-1α is known as a regulator of mitochondrial biogenesis, PGC-1β and ChREBP also cooperatively regulate lipogenesis in liver.7 However, the interaction between PGC-1α and ChREBP has not been studied, and PGC-1α might indirectly control lipogenesis in cancer cells.
In addition to decreased lipogenesis, we also demonstrated higher oxygen consumption and elevated intracellular ATP levels during TGF-β1-induced EMT in A549 cells. Another recent study also showed that mitochondrial ATP production was crucial for cancer cell migration.8 De novo fatty acid synthesis is an energy-consuming pathway, requiring large amounts of acetyl-CoA and NADPH as substrates. Decreased fatty acid synthesis could direct acetyl-CoA toward other metabolic pathways, such as the tricarboxylic acid cycle (TCA cycle) and oxidative phosphorylation, which produce energy very efficiently. This theory is consistent with the higher oxygen consumption noted during TGF-β1-induced EMT, resulting in higher ATP production to fuel cell mobility, elevated migration, and increased metastatic capacity.
At the same time, using theoretical systems biology, Dr. Raphael Levine's laboratory reported energy changes during TGF-β1-induced EMT in A549 lung cancer cells.9 They used single-cell ATP assays to show a dramatically increased cytosolic ATP content during EMT. Their findings are consistent with our conclusions regarding a coordinated energy switch mediated by TGF-β1-induced EMT. Similar to TGF-β1–induced EMT in A549 cells, circulating 4T1 mammary carcinoma cells favored mitochondrial respiration and increased ATP production compared to primary tumors in vivo.6 Thus, we hypothesize that metastatic cells reprogram metabolism from energy-consuming biosynthesis to energy-producing oxidative phosphorylation to support the movement required for metastasis.
More importantly from a therapeutic perspective, like the reversibility of EMT and MET (mesenchymal–epithelial transition), this metabolic switch was also bi-directional. TGF-β1–repressed lipogenic genes, such as ChREBP, were re-expressed once TGF-β1 treatments were withdrawn. This reversion of the metabolic switch might be exploited for prevention of metastasis. This observation also indicates that when metastatic cells finally localize to their new predilected organ site they have the potential to switch their metabolism back to favor growth, resuming their high glycolytic and lipogenic metabolic programs.
Figure 1.
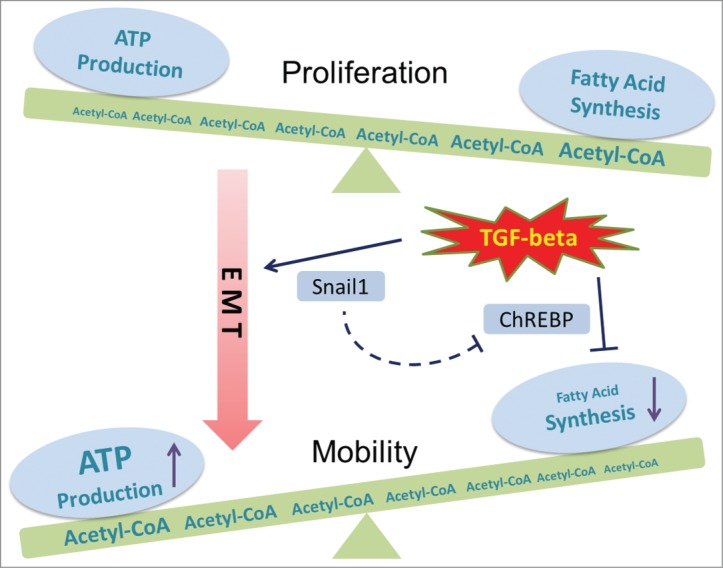
Metabolic regulation during TGF-β1-induced EMT. Transforming growth factor β (TGF-β1) is known to promote epithelial–mesenchymal transition (EMT) through the induction of SNAIL1. Our recent study showed decreased fatty acid synthesis after TGF-β1 treatment, which is regulated through the lipogenic transcriptional factor carbohydrate-responsive element-binding protein (ChREBP). Instead of serving as the substrate for fatty acid synthesis, acetyl-CoA is oxidized through the tricarboxylic acid cycle (TCA cycle) to generate more ATP, which supports the high energy requirement of cell mobility during TGF-β1–induced EMT.
The possibility that decreased fatty acid synthesis regulates and ultimately stimulates cell migration and metastases raises considerable caution regarding the targeting of lipogenesis to cure cancer. We clearly demonstrated that constitutive FASN knockdown was sufficient to induce EMT in the absence of TGF-β1.4 Consistent with previous FASN targeting studies in cancer cells,10 in which the authors stated that FASN could be a potential effective anticancer target, FASN knockdown cells grew more slowly than the non-targeted control cells in culture. However, and most importantly, these same FASN knockdown A549 cells migrated faster both in vitro and in vivo, and ultimately resulted in considerably more death through enhanced lung metastasis in vivo. Thus, although targeting FASN or lipogenesis might suppress growth of primary tumors, a major consequence in our model was enhanced metastasis.
In summary, like most step-wise carcinogenic processes, the growth-suppressing effects of TGF-β1 come with a cost—the stimulation of a metabolic transition that favors migration and metastasis. In addition to induction of an EMT transcriptional program, TGF-β1 also appears to coordinate the metabolic status. The switch from building block accumulation to free energy (ATP) production may help satisfy the energy requirement of migrating cells. Understanding this process at a molecular level should reveal valuable methods of inhibiting cancer progression and metastasis.
References
- 1.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11-20. [DOI] [PubMed] [Google Scholar]
- 2.Currie E, Schulze A, Zechner R, Walther TC, Farese RV, Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013; 18:153-61; PMID:23791484; http://dx.doi.org/ 10.1016/j.cmet.2013.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marin-Valencia I, Cho SK, Rakheja D, Hatanpaa KJ, Kapur P, Mashimo T, Jindal A, Vemireddy V, Good LB, Raisanen J, et al. . Glucose metabolism via the pentose phosphate pathway, glycolysis and Krebs cycle in an orthotopic mouse model of human brain tumors. NMR Biomed 2012; 25:1177-86; PMID:22383401; http://dx.doi.org/ 10.1002/nbm.2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro OM, Deberardinis RJ, Boothman DA. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene 2014; PMID: 25284588; http://dx.doi.org/10655586 10.1038/onc.2014.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2:76-83; PMID:10655586; http://dx.doi.org/ 10.1038/35000025 [DOI] [PubMed] [Google Scholar]
- 6.LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. . PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014; 16:992-1003; PMID:25241037; http://dx.doi.org/ 10.1038/ncb3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambers KT, Chen Z, Lai L, Leone TC, Towle HC, Kralli A, Crawford PA, Finck BN. PGC-1beta and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol Metab 2013; 2:194-204; PMID:24049734; http://dx.doi.org/ 10.1016/j.molmet.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou H, Zhang B, Zheng J, Yu M, Zhou T, Zhao K, Jia Y, Gao X, Chen C, Wei T. The inhibition of migration and invasion of cancer cells by graphene via the impairment of mitochondrial respiration. Biomaterials 2014; 35:1597-607; PMID:24290441; http://dx.doi.org/ 10.1016/j.biomaterials.2013.11.020 [DOI] [PubMed] [Google Scholar]
- 9.Zadran S, Arumugam R, Herschman H, Phelps ME, Levine RD. Surprisal analysis characterizes the free energy time course of cancer cells undergoing epithelial-to-mesenchymal transition. Proc Natl Acad Sci U S A 2014; 111:13235-40; PMID:25157127; http://dx.doi.org/ 10.1073/pnas.1414714111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res 2003; 63:3799-804; PMID:12839976 [PubMed] [Google Scholar]