

Abstract
Glioblastoma multiforme is the most common and most detrimental form of brain tumor, with a current survival time of as little as 14 months. We have recently identified a novel mechanism of therapeutic resistance based on overexpression of the polyamine catabolic enzyme spermidine/spermine N1-acetyltransferase, which promotes DNA repair via chromatin modification.
Keywords: BRCA1, glioblastoma, histone acetylation, SAT1, radiation resistance
Abbreviations
- CNS
central nervous system
- DSB
double-strand break
- GBM
glioblastoma multiforme
- HR
homologous recombination
- MGMT
O6-methylguanine-DNA methyltransferase
- SAT1
spermidine/spermine N1-acetyltransferase
- SSB
single-strand break
- TMZ
temozolomide
Glioblastoma multiforme (GBM) is the most common and most aggressive primary malignant tumor of the central nervous system (CNS). The incidence of GBM increases with age in a bimodal fashion, and peaks in the 6th to 7th decades. The current standard of care has been set by Stupp et al. (1) as maximal safe resection followed by concurrent temozolomide (TMZ) and radiation to a dose of 60 Gy. The favorable arm of the study yielded a median overall survival of 14.6 months, re-emphasizing the dismal prognosis of the disease. GBM is genetically heterogeneous; for example, patients who have tumors with silencing of O6-methylguanine-DNA methyltransferase (MGMT), a DNA repair enzyme, by methylation of its promoter have better survival and a greater response to TMZ (2). Resistance to chemoradiotherapy is the main hurdle in the treatment of GBM. Several mechanisms have been proposed to explain this resistance (3, 4); nevertheless, overcoming it has been elusive. Many therapeutic agents, including ionizing radiation, aim to kill cells through the induction of DNA damage. In order to gain insights into the mechanisms underlying radiation resistance in GBM, our laboratory recently identified the polyamine catabolic enzyme spermidine/spermine N1-acetyltransferase (SAT1) as a principal driver of homologous recombination (HR) repair, an essential pathway for the repair of DNA double-strand breaks (DSBs), through the epigenetic regulation of BRCA1. We found that SAT1 was overexpressed in GBM versus normal brain, and was capable of promoting HR by controlling BRCA1 expression and the formation of BRCA1 foci following damage to DNA. We further demonstrated a novel function of SAT1 as a mediator of histone H3 acetylation controlling BRCA1 gene expression (5). BRCA1 has a critical role in HR, controlling HR foci development, gene expression, and chromatin remodeling (6). Additionally, it has been shown that BRCA1 expression is controlled by the equilibrium between transcriptional co-activators and co-repressors that form a complex and control histone acetylation and DNA accessibility at the BRCA1 promoter (7). Our data demonstrate that the equilibrium between histone acetylation and deacetylation is tipped by SAT1 toward chromatin opening, allowing transcriptional activation of at least the BRCA1 gene (Fig. 1). Aside from chromatin modification affecting gene expression, the relevance of chromatin structure in DNA repair is becoming increasingly clear. Chromatin unpacking and repacking has been recognized as crucial to the assembly of protein complexes at sites of damage, allowing access to DNA to complete repair as well as to signal the end of repair (8). As polyamines are known to pack chromatin (9), we can only speculate at this point that SAT1 plays a role in unpacking sites of damage as an additional facet of its role in DNA repair.
Figure 1.
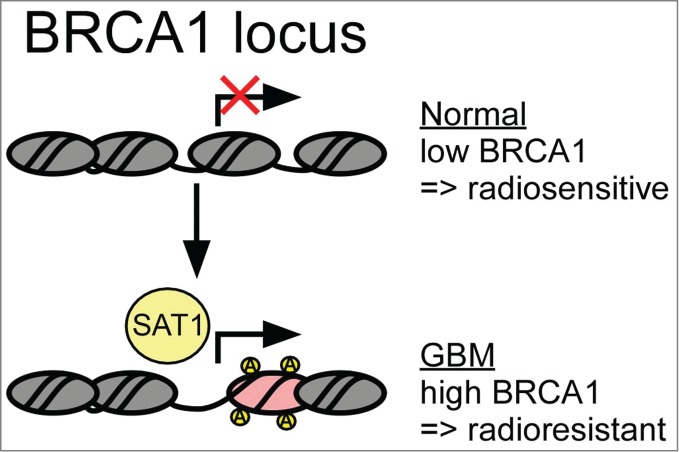
Spermidine/spermine N1-acetyltransferase (SAT1) drives homologous recombination (HR) in glioblastoma multiforme (GBM) by epigenetic control of BRCA1 expression. The BRCA1 locus is known to be regulated by the balance of activating and repressing alterations to histone acetylation. Increased expression of SAT1 in GBM leads to hyperacetylation and activation of BRCA1 expression, and subsequently improved HR and survival following genotoxic stresses such as ionizing radiation.
Our findings carry significance on multiple levels. We have shown that SAT1 knockdown increases radiosensitivity in vitro and in vivo and the immediate clinical implications would be to enhance radiation response by altering SAT1 levels, perhaps via gene therapy or even pharmacologically. However, more specifically, SAT1 depletion led to a dramatic reduction in BRCA1 expression, thus impairing HR; this sparked our interest to capitalize on this effect further by creating more DSBs. One Gy of radiation produces approximately 500–1000 single-strand breaks (SSBs) and 40 DSBs. SSBs are readily repaired; however, with the addition of a poly (ADP-ribose) polymerase (PARP) inhibitor, these SSBs persist into S phase and lead to the creation of more DSBs. Sure enough, the combination of PARP inhibitors with radiation and/or chemotherapy has already been tested with favorable results (10). However, none of the studies evaluating the radio/chemo potentiating effects of PARP inhibitors were in the setting of depleted SAT1 levels (and consequently reduced BRCA1 expression), and thus testing this novel combination may hold great promise to further enhance the effects of radiotherapy. Even though we have demonstrated that SAT1 modulates BRCA1 through the indirect regulation of H3 and have proposed a new role for SAT1 in chromatin regulation suggesting that inhibition of SAT1 may sensitize brain tumors to radiation, there are still many questions to answer. For instance, the effect of SAT1 in regulating the balance of histone acetyl transferases versus histone deactylases in promoter occupancy on the BRCA1 promoter is a natural next step. Additionally, regulating the localized polyamine content at sites of damage is another unexplored area where SAT1 could be a key player, since it has previously been shown that polyamines are involved in DNA and chromatin compaction (9). Overcoming the robust resistance of GBM to therapies is a tall feat, and the combination of multiple approaches is likely necessary in order to achieve respectable results against this resilient disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459-66; PMID:19269895; http://dx.doi.org/ 10.1016/S1470-2045(09)70025-7 [DOI] [PubMed] [Google Scholar]
- 2.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003; PMID:15758010; http://dx.doi.org/ 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- 3.Chakravarti A, Zhai GG, Zhang M, Malhotra R, Latham DE, Delaney MA, Robe P, Nestler U, Song Q, Loeffler J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23:7494-506; PMID:15326475; http://dx.doi.org/ 10.1038/sj.onc.1208049 [DOI] [PubMed] [Google Scholar]
- 4.Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010;12:675-84; PMID:20824044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brett-Morris A, Wright BM, Seo Y, Pasupuleti V, Zhang J, Lu J, Spina R, Bar EE, Gujrati M, Schur R, et al. The polyamine catabolic enzyme SAT1 modulates tumorigenesis and radiation response in GBM. Cancer Res. 2014; 74:6925-34; PMID:25277523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J. The role of BRCA1 in homologous recombination repair in response to replication stress: significance in tumorigenesis and cancer therapy. Cell Biosci. 2013;3:11; PMID:23388117; http://dx.doi.org/ 10.1186/2045-3701-3-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010;17:1406-U500; PMID:21102443; http://dx.doi.org/ 10.1038/nsmb.1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen CC, Tyler J. Chromatin reassembly signals the end of DNA repair. Cell Cycle. 2008;7:3792-7; PMID:19066448; http://dx.doi.org/ 10.4161/cc.7.24.7188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warters RL, Newton GL, Olive PL, Fahey RC. Radioprotection of human cell nuclear DNA by polyamines: radiosensitivity of chromatin is influenced by tightly bound spermine. Radiat Res. 1999;151:354-62; PMID:10073674; http://dx.doi.org/ 10.2307/3579948 [DOI] [PubMed] [Google Scholar]
- 10.Barazzuol L, Jena R, Burnet NG, Meira LB, Jeynes JC, Kirkby KJ, Kirkby NF. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat Oncol. 2013;8:65; PMID:23510353; http://dx.doi.org/ 10.1186/1748-717X-8-65 [DOI] [PMC free article] [PubMed] [Google Scholar]