

Abstract
Autophagy is an evolutionarily conserved intracellular catabolic process that is used by all cells to degrade dysfunctional or unnecessary cytoplasmic components through delivery to the lysosome. Increasing evidence reveals that autophagic dysfunction is associated with human diseases, such as cancer. Paradoxically, although autophagy is well recognized as a cell survival process that promotes tumor development, it can also participate in a caspase-independent form of programmed cell death. Induction of autophagic cell death by some anticancer agents highlights the potential of this process as a cancer treatment modality. Here, we review our current understanding of the molecular mechanism of autophagy and the potential roles of autophagy in cell death, cancer development, and cancer treatment.
Keywords: autophagy, apoptosis, Atg gene, caspase, necrosis, programmed cell death
Abbreviations
- AMPK
AMP-activated protein kinase
- APC
adenomatous polyposis coli
- APF
after puparium formation
- Atg
autophagy-related gene
- BRCA1, breast cancer 1
early onset
- BRAF
V-Raf Murine Sarcoma Viral Oncogene Homolog B
- DAPK
death-associated protein kinase
- Ecdysone
20-hydroxyecdysone
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- IP3
Inositol-1,4,5 trisphosphate
- LC3
microtubule-associated protein light chain 3
- MEF
mouse embryonic fibroblast
- Min
multiple intestinal neoplasia
- MPP+
1-methyl-4-phenylpyridinium ion
- mTOR
mechanistic target of rapamycin
- PAS
pre-autophagosomal structure
- PDAC
pancreatic ductal adenocarcinoma
- PE
phosphatidylethanolamine
- PI(3)K
phosphatidylinositol 3-kinase
- PtdIns
phosphatidylinositol
- PtdIns(3)P
phosphatidylinositol-3-phosphate
- PtdIns(3,5)P2
phosphatidylinositol 3,5-bisphosphate
Introduction
Like cell growth and division, programmed cell death plays a fundamental role in tissue and organism homeostasis.1,2 Three types of programmed cell death occur during development—apoptosis, autophagic cell death, and necrosis.3 Abnormal regulation of programmed cell death is associated with a wide variety of human diseases, including cancer. Although the contribution of a loss of apoptotic responses to tumor development is established and has been extensively analyzed at the molecular level, much less is known about the mechanisms that regulate autophagic cell death and how this may contribute to cancer.
Autophagy is a general term for the process by which cytoplasmic material is delivered to lysosomes for degradation. There are 3 types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy is the focus of this review. During macroautophagy (hereafter simply called autophagy), an isolation membrane encloses a small portion of cytoplasmic material, including damaged organelles and unused proteins, to form a double-membraned structure called an “autophagosome” (Fig. 1). The outer membrane of an autophagosome subsequently fuses with the membrane of lysosomes to become an “autolysosome”, in which the cytoplasmic material is degraded by lysosomal enzymes.
Figure 1.
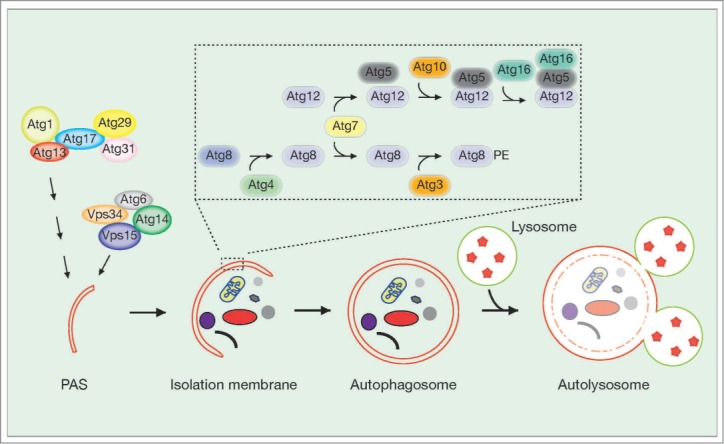
Autophagy regulatory pathway. After autophagy induction, the Atg1 complex (Atg1–Atg13–Atg17–Atg29–Atg31) translocates to the endoplasmic reticulum (ER), which is thought to be the major membrane source for autophagy (other membrane sources may include mitochondria and the plasma membrane). This leads to recruitment of the autophagy-specific form of the phosphatidylinositol 3-kinase (PI(3)K) complex, which includes Vps34, Vps15, Atg6/Beclin-1 and Atg14, to the ER. To form an autophagosome, elongation and closure of the isolation membrane requires 2 protein conjugation systems, the Atg12–Atg5–Atg16 complex and the Atg8/LC3–phosphatidylethanolamine (PE) complex. See text for more details.
Analyses of autophagy-defective organisms have revealed numerous physiological and pathological roles of autophagy at both the cellular and organismal levels. This review focuses on the molecular control of autophagy and its possible functions in programmed cell death. We will also discuss the potential role of autophagic cell death in cancer therapy.
The Core Autophagy Machinery
The genes that regulate autophagy were first identified in yeast,4-7 although most of these factors are conserved in higher eukaryotes, including humans (Fig. 1).8 To date, more than 30 autophagy-related (Atg) genes have been reported in yeast.9 Upon the induction of autophagy, Atg1/ULK1 kinase and its complex components Atg13, Atg17/FIP200, Atg29, and Atg31 translocate to the pre-autophagosomal structure (PAS).9-11 This leads to recruitment of the autophagy-specific form of the phosphatidylinositol (PtdIns) 3-kinase (PI(3)K) complex, which includes Vps34, Vps15, Atg6/Beclin-1, and Atg14, to the PAS.12,13 The PI(3)K complex produces phosphatidylinositol-3-phosphate (PtdIns(3)P), which recruits effector proteins such as Atg18/WIPI1/2 to the PAS. Atg18 forms a complex with Atg2 that functions in autophagosome formation,14 and also controls the size of vesicles and phosphatidylinositol 3,5-bisphosphate (PtdIns (3, 5) P2) homeostasis in complex with other proteins.15 At the final step of autophagosome formation, elongation and closure of the isolation membrane requires 2 protein conjugation systems, the Atg12–Atg5–Atg16 complex16 and the Atg8/LC3–phosphatidylethanolamine (PE) complex.17 The ubiquitin-like Atg12 protein is conjugated with Atg5 by Atg7 (E1-like) and Atg10 (E2-like) enzymes, and then Atg12-Atg5 conjugate further to interact with Atg16 and function as a complex.18-20 Atg8 is first processed by the protease Atg4, and is conjugated with PE by the Atg7 and Atg3 (E2-like) enzymes.17 Biochemical evidence supports a model in which the Atg12–Atg5 complex possesses an E3-like activity for efficient PE lipidation of Atg8.21
It is generally believed that all of the core machinery proteins are essential for autophagosome formation. However, several recent findings indicate that autophagy can proceed without some of the Atg proteins. Mouse embryonic fibroblast (MEF) cells from either Atg5-/- or Atg7-/- knockout mice formed autophagosomes and autolysosomes, and performed autophagy-regulated protein degradation.22 However, lipidation of the microtubule-associated protein light chain 3 (LC3, the mammalian homolog of yeast Atg8) did not occur during this Atg5/Atg7-independent autophagy.22 During the same year, Chu and colleagues reported that the parkinsonian neurotoxin MPP+ induces autophagy and mitochondrial degradation independent of Beclin-1.23 In addition, Chang and colleagues showed that loss of either Atg7 or Atg3 function fails to influence the autophagy that participates in programmed reduction of cell size during Drosophila intestine cell death.24 These studies indicate that autophagy can be controlled by different pathways in a cell context- and organism-specific manner.
In addition to their role in regulating autophagy activity, there is increasing evidence indicating that Atg proteins also have non-autophagic biological functions.25 Atg6/Beclin-1 has been reported to function as a tumor suppressor, and Beclin-1+/– tumors in mice possess elevated cell stress and p62 levels, altered NF-κB signaling, and genome instability.26 Drosophila lacking Atg6 function exhibit blood cell tumors, but also possess defects in multiple vesicle trafficking pathways.27 Therefore, it is possible that, in addition to autophagy, altered endocytosis and protein secretion may contribute to tumor development. Eukaryotic cells release proteins into the extracellular space by 2 main routes. One is the conventional secretion pathway for proteins that contain a signal for translocation into the endoplasmic reticulum (ER), which is followed by their vesicular transport to Golgi membranes and subsequent export from the cell. The second is the unconventional secretion pathway for proteins that lack a secretion signal for entry into the ER–Golgi membrane pathway. Examples of proteins that use this secretory pathway include acyl-CoA-binding protein and the cytokines interleukin 1-β and interleukin-6. The mechanisms underlying the unconventional protein secretion pathway are poorly understood, but evidence indicates that these proteins are secreted by an autophagosome-like vesicular intermediate that requires Atg proteins such as Atg5, Atg7, and Atg12.28-30 Unlike autophagy, these vesicles fuse with the plasma membrane, and bypass the final stages of autophagy.
Regulators of Autophagy
Autophagy is a tightly regulated pathway that can be induced by a variety of stimuli, such as nutrient deprivation, hypoxia, reactive oxygen species, protein aggregates, and damaged organelles. The activation of autophagy by these stimuli involves multiple signaling pathways. For example, the mechanistic target of rapamycin (mTOR), a serine/threonine protein kinase, negatively regulates the activation of autophagy. In nutrient-rich conditions, mTOR interacts with Atg13 and phosphorylates it at several serine residues. The phosphorylation of Atg13 reduces both its affinity for Atg1/ULK1 and Atg1/ULK1 activity. Upon mTOR inhibition, for example by starvation, mTOR phosphorylation of Atg13 is reduced, enabling activation of Atg1/ULK1 kinase activity and autophagy.10
Antiapoptotic proteins, such as Bcl-2, Bcl-XL, and Mcl-1, have also been proposed as important negative regulators of autophagy. These proteins bind to the BH3 domain of Beclin-1 through their BH3-binding groove, and inhibit Beclin-1–dependent autophagy induction.31,32 Proapoptotic BH3-only proteins and pharmacological BH3 mimetics can induce autophagy by competitively disrupting the interaction between Beclin-1 and Bcl-2 or Bcl-XL.32 In addition, death-associated protein kinase (DAPK) can promote dissociation of Beclin-1 from Bcl-XL and induce autophagy by mediating phosphorylation of the BH3 domain of Beclin-1.33 However, it is important to note that the role of Bcl-2 in the regulation of autophagy is a subject of debate. For example, there is evidence indicating that the effect of prosurvival Bcl-2 family members on autophagy is instead an indirect consequence of their inhibition of the apoptosis mediators Bcl-2-associated X (Bax) and Bcl-2 homologous antagonist/killer (Bak). In the absence of Bax and Bak, antagonizing or altering the levels of prosurvival Bcl-2 family members has no detectable impact on autophagy.34
Inositol-1,4,5 trisphosphate (IP3) is a secondary messenger molecule that mediates calcium release from the ER by binding the IP3 receptor, which is an ER-localized calcium ion channel.35 Increasing evidence shows that IP3 signaling pathway components, including the IP3 receptor, IP3 kinase 2, and calmodulin, are involved in regulating autophagy.36,37 An increase in the level of free cytosolic calcium also triggers autophagy. This process is mediated by calcium/calmodulin-dependent kinase kinase-β and AMP-activated protein kinase (AMPK) via mTOR inhibition.38 In addition to inhibition of mTOR, AMPK directly interacts with and phosphorylates Atg1 to influence autophagy.39,40
Steroid hormone has also been shown to regulate autophagy activity, and this has been best studied during development in Drosophila melanogaster. In Drosophila, pulses of the steroid 20-hydroxyecdysone (ecdysone) control the transitions through different developmental stages.41 At the end of the third larval instar stage, an ecdysone pulse triggers formation of the prepupa. The increase in ecdysone at this stage activates autophagy in the larval midgut, and this autophagy promotes intestine cell death.42 Ten hours later, the subsequent increase in ecdysone triggers formation of the pupa, and activates autophagy that promotes programmed cell death of the salivary gland.43 The molecular mechanisms underlying this process have been described extensively elsewhere.44,45
Autophagy in Cell Death
Programmed cell death is a conserved and genetically regulated process that plays important roles throughout the lives of metazoans. Schweichel and Merker identified 3 types of programmed cell death based on morphology: apoptosis, autophagic cell death, and necrosis.3 Among them, autophagic cell death is characterized by the presence of abundant autophagosomes in the dying cell and the lack of phagocyte participation in cell death. However, whether autophagy is the mechanism by which cells actually die (cell death by autophagy) or is simply present during cell death (cell death with autophagy) has been a subject of controversy because autophagy is well recognized as a cell survival mechanism.46-48 During conditions of nutrient limitation, autophagy is used to generate amino acids and energy to maintain cell viability through the bulk degradation of cytoplasmic material. Accordingly, the presence of autophagy in dying cells has been proposed to be a stress response mechanism to prolong cell viability.
Nevertheless, recent studies strongly support autophagy as a process that can promote programmed cell death. The contribution of autophagy to cell death has been studied most extensively in Drosophila. As described above, an increase in steroid hormone levels triggers the destruction of larval tissues during the transition from a larva to an adult.41 Destruction of the larval salivary gland requires both autophagy and caspase activities.45,49 Mutations in either multiple Atg genes or caspase genes, or overexpression of the caspase inhibitor p35, lead to incomplete degradation of larval salivary glands. However, combined inhibition of both autophagy and caspase activities increases suppression of salivary gland degradation. Further, Atg1-triggered autophagy in salivary glands is sufficient to induce premature cell death in a caspase-independent manner.45 These data indicate that caspases and autophagy function additively in the degradation of Drosophila larval salivary glands.
In contrast to the Drosophila salivary gland, death of the larval midgut cells of the intestine is not disrupted by overexpression of p35 or by mutation of caspases, indicating that the canonical apoptosis pathway is not required for developmental midgut cell death.50 Interestingly, the impaired function of multiple Atg genes, including either Atg1, Atg2 or Atg18, blocks larval midgut degradation.50 Additionally, caspase deficiency fails to enhance the Atg mutant phenotype in the midgut. These data indicate that autophagy, and not apoptosis pathway components, is essential for Drosophila midgut programmed cell death.
Studies in mammalian systems also provide evidence in support of concurrent activation of autophagic and apoptotic pathways. In U937 monocytoid cells and L929 fibrosarcoma cells, knockdown of either Beclin-1 or Atg7, 2 essential Atg genes, blocks non-apoptotic cell death induced by caspase-8 inhibition.51 In response to death stimulation, Bax-/-Bak-/- double knockout MEFs undergo non-apoptotic cell death. This cell death is associated with increased numbers of autophagosomes and autolysosomes, and can be reduced by knockdown of either Atg5 or Beclin-1.52 In human ovarian surface epithelial cells, expression of oncogenic H-RasV12 leads to caspase-independent cell death with features of autophagy. This Ras-induced autophagy-dependent cell death was associated with upregulation of the BH3-only protein Noxa and the autophagy regulator Beclin-1.53 Furthermore, it has been reported that activation of autophagy by the autophagy-inducing peptide Tat-Beclin-1 can cause cell death with unique morphological features of autophagy. This type of cell death is blocked by either pharmacological or genetic inhibition of autophagy, but not by impairment of known regulators of either apoptosis or necroptosis.54 In neonatal mice, neuron-specific deletion of Atg7 protects against cerebral hypoxia–ischemia-induced hippocampal neuron death,55 and in adult rats shRNA targeting Beclin-1 prevents neuronal death in the thalamus following focal cerebral infarction.56 Although such studies provide genetic data in support of autophagy function in the death of mammalian cells and tissues, additional analyses of autophagy in developmental cell death are needed in mammalian model systems.
Autophagy might be required for cell death, but little is known about how autophagy kills cells. One possibility is that autophagy causes a metabolic catastrophe by depleting mitochondria and metabolic substrates. Studies in Drosophila provide some support for this model, since high levels of autophagy that are induced by Atg1 expression are sufficient to induce cell death that is either dependent or independent of caspase function, depending on cell type.24,45,57 Another possible mechanism for autophagy-dependent cell death is the selective recruitment of cell survival factors to autophagosomes for degradation. Studies in mammalian cells have shown that selective recruitment of cytoplasmic catalase to autophagosomes leads to accumulation of reactive oxygen species and cell death.58 Similarly, recruitment of the inhibitor of apoptosis Bruce to autophagosomes in the Drosophila ovary leads to the activation of caspases and cell death.59 Finally, it has been proposed that autophagic membrane structures could serve as signaling scaffolds to enable activation of either apoptotic or programmed necrosis protein complexes. Limited data exist in support of each of these possible mechanisms, and more work is needed to determine how autophagy promotes cell death.
Cell-Autonomous Regulation of Autophagy
Autophagy, which literally means “self-eating," is involved in the bulk degradation of long-lived cytosolic proteins and organelles, and is an important intracellular biomass quality and quantity control process. All of the autophagy genes and proteins identified to date function in a cell-autonomous manner. In yeast, for example, nutrient deprivation induces a high level of autophagy, which provides a cell-autonomous source (by autodigestion of the cytosol) of energy and amino acids for the synthesis of proteins that are essential for survival. In the Drosophila fat body, which is a nutrient storage organ, clonal knockdown of Atg5 inhibits starvation-induced autophagy only in cells with reduced autophagy gene function.60 Similarly, clonal loss of Atg1 function in dying Drosophila midgut cells specifically inhibits autophagy in cells that possess reduced autophagy gene function (Fig. 2).24 Draper, the Drosophila ortholog of the Caenorhabditis elegans engulfment receptor CED-1, is required for autophagy activation during larval salivary gland developmental cell death.61 Loss of draper prevents the induction of autophagy, and causes an incomplete larval salivary gland degradation phenotype. Interestingly, unlike its function in phagocytosis, Draper also regulates autophagy in a cell-autonomous manner.61 Knockdown of draper does not prevent starvation-induced autophagy in the fat body, indicating that regulation of autophagy by Draper is also tissue-specific. As Draper is an engulfment receptor, it is commonly accepted that there is a corresponding ligand; however, the ligand required for the regulation of autophagy by Draper remains unknown. If Draper does require a ligand for the regulation of autophagy, it is possible that this ligand functions in a non–cell-autonomous manner.
Figure 2.

Cell-autonomous function of autophagy genes. (A) Experimental design. A cell with Atg gene knockdown by RNAi co-expresses green fluorescent protein (GFP, green) and is surrounded by wild type cells (white). After autophagy induction, red autophagy reporter punctae are present only in wild type cells, and not in the Atg RNAi knockdown cell. The Atg RNAi knockdown cell is larger than wild type cells, because autophagy leads to a reduction in cell size. (B) An image of the mid gut of the Drosophila larval intestine. Cells with Atg1 knockdown are marked by GFP (green). mCherry-tagged Atg8a protein (red), which localizes to autophagosomes and autolysosomes, is expressed in all cells and serves as a reporter of autophagy. Two hours after puparium formation (APF), mCherry-Atg8a punctae are only present in the wild type mid gut cells, but not in the Atg1 RNAi-expressing green cells. Scale bar represents 50 μm. Image courtesy of T.-K. Chang.
Role of Autophagy in Cancer Development
Cancer was one of the first human disorders that was linked to a defect in autophagy.62,63 However, the role of autophagy in tumor progression is still an enigma. Inactivation of Fip200, the mouse homolog of yeast Atg17, in the polyoma middle T (PyMT) mouse mammary cancer model impairs tumor growth.64 Deletion of either Atg5 or Atg7 in the mouse liver causes hepatoma formation without progression to hepatocellular carcinoma.65 Downregulation of essential autophagy proteins abrogates the tumorigenicity of oncogenic RAS-expressing human and mouse cancer cell lines.66,67 These findings suggest that autophagy can be tumor promoting in established cancers. It has been suggested that, through intracellular recycling, autophagy provides substrates that enable tumor cells to survive the metabolic stress in the tumor microenvironment and promotes tumor progression.
Paradoxically, other studies support a role for autophagy in tumor suppression. Mice with allelic loss of Beclin-1 have decreased autophagy and are more prone to the development of spontaneous tumors, including lymphomas, lung carcinomas, hepatocellular carcinomas, and mammary precancerous lesions.68,69 In addition, immortalized kidney and mammary epithelial cells derived from Beclin-1 heterozygous-deficient mice showed increased tumorigenicity when transplanted into immunocompromised mice.70,71 Atg5 is frequently downregulated in primary melanomas compared to benign nevi, and analyses of 158 patient biopsies showed that patients with low levels of Atg5 in their tumors had reduced progression-free survival.72 The mechanisms by which autophagy functions in tumor suppression remain unclear, and it is possible that autophagy could directly restrict cell proliferation by inducing cell death. It is important to note that Laddha and colleges recently found that most Beclin-1 mutations are associated with mutations in the BRCA1 tumor suppressor gene.73 Although the ability to draw strong conclusions may be limited by the current availability of cancer genome data, these data do raise doubts about the tumor suppressor function of Beclin-1.
Increasing evidence suggests that autophagy has a specific influence on tumor progression depending on context. Inactivation of either Atg5 or Atg7 impairs the initiation of KrasG12D-driven lung cancer, and deletion of the tumor suppressor p53 restores cancer progression in either Kras Atg5-/- or Kras Atg7-/- tumors.74 In a humanized genetically modified mouse model of Kras-triggered pancreatic ductal adenocarcinoma (PDAC), a small number of precancerous lesions developed into PDAC over time. If these mice also lacked either the Atg5 or Atg7 autophagy gene, they accumulated low-grade, premalignant pancreatic intraepithelial neoplastic lesions, and progression to high-grade pancreatic intraepithelial neoplasias and PDAC was blocked. In contrast, in mice containing oncogenic Kras and lacking p53, loss of autophagy no longer blocked tumor progression and actually accelerated tumor onset.75 However, decreased p53 function had no impact on the influence of autophagy inhibition on tumor growth in a different study of PDAC.76 The discrepancy between these studies may indicate that the timing of p53 impairment influences the impact of autophagy modulation on tumor growth. Furthermore, a recent study shows that the influence of autophagy on tissue overgrowth depends on the growth-inducing stimulus and cell type.77 These findings support the notion that autophagy may play distinct roles in cancer as both an inhibitor of initial oncogenesis and then as a facilitator of tumor progression, and that cell and tumor context influence how autophagy impacts tumor development. These studies have important implications for the use of modulators of autophagy for cancer therapy.
Autophagy and Cell Death in Response to Anticancer Agents
Autophagy has been reported to play contradictory roles in tumor initiation and progression. Therefore, both repression and stimulation of autophagy should to be considered as therapeutic approaches depending on the many factors that contribute to cancer. Therapeutic induction of autophagy-associated cell death can be accomplished by modulation of regulators of autophagy. mTOR is a key regulator of cell growth and an autophagy repressor, and inhibition of this kinase leads to the activation of Atg1/Ulk1 and stimulation of autophagy. Treatment with the mTOR inhibitor rapamycin led to a reduction in carcinogen-induced lung tumors in a murine model.78 In addition, rapamycin treatment showed antitumor effects in the MCF7 and MDA-MB-231 breast cancer cell lines in xenografted tumors, and it was suggested that this was because of inhibition of angiogenesis.79 Furthermore, continuous long-term rapamycin treatment in APCMin/+ mice, which have enhanced AKT-mTOR signaling, was shown to markedly inhibit intestinal neoplasia.80 Recently, combining autophagy activation by mTOR inhibition with radiation was shown to lead to enhanced therapeutic effects in cancer cells and xenografts.81 Another strategy that has been implemented is the induction of autophagy by inhibition of mTOR while impairing lysosome function by treatment with chloroquine.82 This leads to the accumulation of what is presumably toxic autophagic cargo without the lysosomal capacity to degrade this material and thus results in cell death.
As described earlier, autophagy can also be induced by the protein kinase AMPK. Metformin is an inhibitor of the mitochondrial electron transport chain complex I that leads to decreased ATP production and increased levels of AMP. This causes AMPK activation and autophagy induction. Metformin has been reported to prevent tobacco carcinogen-induced lung tumorigenesis in a rodent cancer model,83 and to improve tumor oxygenation and hence the radiotherapy response.84 Furthermore, the combination of chemotherapy and metformin is more harmful to breast cancer cells than treatment with chemotherapy or metformin alone.85 Significantly, the first clinical trial that combined autophagy inhibition and chemotherapy demonstrated that patients harboring the BRAFV600E mutation who were treated with the RAF inhibitor vemurafenib and chloroquine had decreased brain tumor growth.86 Although the contribution of autophagy to these cancer therapeutic strategies is not completely clear, the results of this clinical trial illustrate how targeting autophagy could lead to new cancer therapies.
Growing evidence indicates that autophagy not only preserves cellular homeostasis in conditions of endogenous stress, but also plays an important role in controlling intracellular pathogens. Thus, autophagy represents one of the most primitive innate immune responses. From the immunological point of view, cancer can only develop when premalignant cells escape immunosurveillance by either losing their antigenic properties or by actively suppressing the antitumor immune response.87 Autophagy is often thought to be suppressed in tumor cells during early oncogenesis, such as upon allelic loss of Beclin-1. Indeed, autophagy-deficient tumors fail to elicit an anticancer immune response upon exposure to chemotherapy.88 Many preclinical studies of the role of autophagy in killing tumor cells may have missed important non-autonomous influences on the immune system, since previous work has mostly been conducted either in vitro or in animal models with defective immune systems. Therefore, when considering modulation of autophagy for cancer therapy, it is important to consider the role of autophagy in the immune and anticancer responses.
Conclusions and Future Directions
Organisms continuously replace their damaged cells to maintain health and to make morphological and functional changes via programmed cell death. As the most studied form of programmed cell death, analyses of apoptosis have led to important contributions to our understanding of cancer biology and have provided potential targets for new cancer therapies. However, the apoptotic machinery is often mutated in human tumors, and modulation of alternative forms of programmed cell death may provide important alternatives for cancer treatment. Here, we have discussed the molecular mechanisms of autophagy, how autophagy may contribute to cell death, and its potential role in cancer. However, there are many fundamental questions that remain to be addressed. For example, although studies of invertebrate organisms have made important contributions to our understanding of the role of autophagy in cell death, the physiological role of autophagic cell death in mammalian organisms needs to be examined further. Additionally, the molecular mechanisms that specifically regulate autophagic cell death are still far from understood. A better characterization of these mechanisms will help us to specifically induce the cell death function of autophagy while inhibiting its cell survival function in tumor cells.
Critical issues remain when considering autophagic cell death in disease therapeutics. One challenge in the study of cancer treatment is the fact that autophagy has dual roles in tumorigenesis. Both stimulation and repression of autophagy could be promising approaches, but existing drugs that influence autophagy probably influence other processes, including multiple vesicle trafficking pathways. Therefore, the development of drugs that specifically and exclusively stimulate or repress autophagy is very important. Autophagy mitigates stress in both target tumor and healthy normal cells, and it is important to consider how to design drugs that specifically target tumor cells without damaging neighboring normal tissue. Likewise, cancer is a very complicated disease; different tumor types and different patient genotypes may vary in their responses to autophagy modulation, and therefore may differ in sensitivity to the modulation of autophagy as a therapeutic strategy. Thus, a better understanding of the influence of different cell types, genotypes, and environmental factors on the response of tumors to the modulation of autophagy is essential in order to understand the potential of manipulating autophagy for cancer therapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Acknowledgments
We thank T.-K. Chang for images.
Funding
Research related to this subject is supported by NIH grants GM079431, CA159314 and AI099708. E.H.B. is an Ellison Medical Foundation Scholar.
References
- 1.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell 2011; 147:742-58; PMID:22078876; http://dx.doi.org/ 10.1016/j.cell.2011.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lockshin RA, Williams CM. Programmed cell death-I. Cytology of degeneration in the intersegmental muscles of the pernyi silkmoth. J Insect Physiol 1965; 11:123-33; PMID:14287218; http://dx.doi.org/ 10.1016/0022-1910(65)90099-5 [DOI] [PubMed] [Google Scholar]
- 3.Schweichel J-U, Merker H-J. The morphology of various types of cell death in prenatal tissues. Teratology 1973; 7:253-66; PMID:4807128; http://dx.doi.org/ 10.1002/tera.1420070306 [DOI] [PubMed] [Google Scholar]
- 4.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993; 333:169-74; PMID:8224160; http://dx.doi.org/ 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- 5.Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, Wolf DH. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett 1994; 349:275-80; PMID:8050581; http://dx.doi.org/ 10.1016/0014-5793(94)00672-5 [DOI] [PubMed] [Google Scholar]
- 6.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Bio 1995; 131:591-602; PMID:7593182; http://dx.doi.org/ 10.1083/jcb.131.3.591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harding TM, Hefner-Gravink A, Thumm M, Klionsky DJ. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein. J Biol Chem 1996; 271:17621-4; PMID:8663607; http://dx.doi.org/ 10.1074/jbc.271.30.17621 [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/ 10.1016/j.cell.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 9.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2010; 80:125-56; PMID:21548784; http://dx.doi.org/ 10.1146/annurev-biochem-052709-094552 [DOI] [PubMed] [Google Scholar]
- 10.Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507-13; PMID:10995454; http://dx.doi.org/ 10.1083/jcb.150.6.1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997; 192:245-50; PMID:9224897; http://dx.doi.org/ 10.1016/S0378-1119(97)00084-X [DOI] [PubMed] [Google Scholar]
- 12.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 2001; 152:519-30; PMID:11157979; http://dx.doi.org/ 10.1083/jcb.152.3.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009; 186:773-82; PMID:19797076; http://dx.doi.org/ 10.1083/jcb.200907014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obara K, Sekito T, Niimi K, Ohsumi Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem 2008; 283:23972-80; PMID:18586673; http://dx.doi.org/ 10.1074/jbc.M803180200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Efe JA, Botelho RJ, Emr SD. Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol Biol Cell 2007; 18:4232-44; PMID:17699591; http://dx.doi.org/ 10.1091/mbc.E07-04-0301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395:395-8; PMID:9759731; http://dx.doi.org/ 10.1038/26506 [DOI] [PubMed] [Google Scholar]
- 17.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488-92; PMID:11100732; http://dx.doi.org/ 10.1038/35044114 [DOI] [PubMed] [Google Scholar]
- 18.Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, Kominami E. Apg7p/Cvt2p: A novel protein-activating enzyme essential for autophagy. Mol Biol Cell 1999; 10:1367-79; PMID:10233150; http://dx.doi.org/ 10.1091/mbc.10.5.1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J 1999; 18:5234-41; PMID:10508157; http://dx.doi.org/ 10.1093/emboj/18.19.5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5. Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 2002; 277:18619-25; PMID:11897782; http://dx.doi.org/ 10.1074/jbc.M111889200 [DOI] [PubMed] [Google Scholar]
- 21.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298-302; PMID:17986448; http://dx.doi.org/ 10.1074/jbc.C700195200 [DOI] [PubMed] [Google Scholar]
- 22.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654-8; PMID:19794493; http://dx.doi.org/ 10.1038/nature08455 [DOI] [PubMed] [Google Scholar]
- 23.Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 2007; 3:663-6; PMID:17622797; http://dx.doi.org/ 10.4161/auto.4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang T-K, Shravage BV, Hayes SD, Powers CM, Simin RT, Harper JW, Baehrecke EH. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol 2013; 15:1067-78; PMID:23873149; http://dx.doi.org/ 10.1038/ncb2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep 2013; 14:143-51; PMID:23337627; http://dx.doi.org/ 10.1038/embor.2012.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062-75; PMID:19524509; http://dx.doi.org/ 10.1016/j.cell.2009.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shravage BV, Hill JH, Powers CM, Wu L, Baehrecke EH. Atg6 is required for multiple vesicle trafficking pathways and hematopoiesis in Drosophila. Development 2013; 140:1321-9; PMID:23406899; http://dx.doi.org/ 10.1242/dev.089490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol 2010; 188:537-46; PMID:20156962; http://dx.doi.org/ 10.1083/jcb.200911149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J 2011; 30:4701-11; PMID:22068051; http://dx.doi.org/ 10.1038/emboj.2011.398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov 2014; 4:466-79; PMID:24513958; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 32.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527-39; PMID:17446862; http://dx.doi.org/ 10.1038/sj.emboj.7601689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/ 10.1038/embor.2008.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindqvist LM, Heinlein M, Huang DC, Vaux DL. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci U S A 2014; 111:8512-7; PMID:24912196; http://dx.doi.org/ 10.1073/pnas.1406425111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000; 1:11-21; PMID:11413485; http://dx.doi.org/ 10.1038/35036035 [DOI] [PubMed] [Google Scholar]
- 36.Nelson C, Ambros V, Baehrecke EH. miR-14 regulates autophagy during developmental cell death by targeting ip3-kinase 2. Mol Cell 2014; 56(3):376-88; PMID:25306920; http://dx.doi.org/ 10.1016/j.molcel.2014.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16:1006-17; PMID:19325567; http://dx.doi.org/ 10.1038/cdd.2009.34 [DOI] [PubMed] [Google Scholar]
- 38.Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25:193-205; PMID:17244528; http://dx.doi.org/ 10.1016/j.molcel.2006.12.009 [DOI] [PubMed] [Google Scholar]
- 39.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/ 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang C, Baehrecke EH, Thummel CS. Steroid regulated programmed cell death during Drosophila metamorphosis. Development 1997; 124:4673-83; PMID:9409683 [DOI] [PubMed] [Google Scholar]
- 42.Lee C-Y, Cooksey BAK, Baehrecke EH. Steroid regulation of midgut cell death during Drosophila development. Dev Biol 2002; 250:101-11; PMID:12297099; http://dx.doi.org/ 10.1006/dbio.2002.0784 [DOI] [PubMed] [Google Scholar]
- 43.Lee C-Y, Baehrecke EH. Steroid regulation of autophagic programmed cell death during development. Development 2001; 128:1443-55; PMID:11262243 [DOI] [PubMed] [Google Scholar]
- 44.Lee C-Y, Simon CR, Woodard CT, Baehrecke EH. Genetic mechanism for the stage- and tissue-specific regulation of steroid-triggered programmed cell death in Drosophila. Dev Biol 2002; 252:138-48; PMID:12453466; http://dx.doi.org/ 10.1006/dbio.2002.0838 [DOI] [PubMed] [Google Scholar]
- 45.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131:1137-48; PMID:18083103; http://dx.doi.org/ 10.1016/j.cell.2007.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 2005; 6:505-10; PMID:15928714; http://dx.doi.org/ 10.1038/nrm1666 [DOI] [PubMed] [Google Scholar]
- 47.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115:2679-88; PMID:16200202; http://dx.doi.org/ 10.1172/JCI26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15:81-94; PMID:24401948; http://dx.doi.org/ 10.1038/nrm3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin DN, Baehrecke EH. Caspases function in autophagic cell death in Drosophila. Development 2004; 131:275-84; PMID:14668412; http://dx.doi.org/ 10.1242/dev.00933 [DOI] [PubMed] [Google Scholar]
- 50.Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol 2009; 19:1741-6; PMID:19818615; http://dx.doi.org/ 10.1016/j.cub.2009.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500-2; PMID:15131264; http://dx.doi.org/ 10.1126/science.1096645 [DOI] [PubMed] [Google Scholar]
- 52.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nature Cell Biol 2004; 6:1221-8; PMID:15558033; http://dx.doi.org/ 10.1038/ncb1192 [DOI] [PubMed] [Google Scholar]
- 53.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011; 42:23-35; PMID:21353614; http://dx.doi.org/ 10.1016/j.molcel.2011.02.009 [DOI] [PubMed] [Google Scholar]
- 54.Liu Y, Shoji-Kawata S, Sumpter RMJ, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 2013; 110:20364-71; PMID:24277826; http://dx.doi.org/ 10.1073/pnas.1319661110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol 2008; 172:454-69; PMID:18187572; http://dx.doi.org/ 10.2353/ajpath.2008.070876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xing S, Zhang Y, Li J, Zhang J, Li Y, Dang C, Li C, Fan Y, Yu J, Pei Z, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 2012; 8:63-76; PMID:22108007; http://dx.doi.org/ 10.4161/auto.8.1.18217 [DOI] [PubMed] [Google Scholar]
- 57.Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol 2007; 17:1-11; PMID:17208179; http://dx.doi.org/ 10.1016/j.cub.2006.10.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo MJ. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 2006; 103:4952-7; PMID:16547133; http://dx.doi.org/ 10.1073/pnas.0511288103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjørkøy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, Stenmark H. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol 2010; 190:523-31; PMID:20713604; http://dx.doi.org/ 10.1083/jcb.201002035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004; 7:167-78; PMID:15296714; http://dx.doi.org/ 10.1016/j.devcel.2004.07.009 [DOI] [PubMed] [Google Scholar]
- 61.McPhee CK, Logan MA, Freeman MR, Baehrecke EH. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature 2010; 465:1093-6; PMID:20577216; http://dx.doi.org/ 10.1038/nature09127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009; 15:5308-16; PMID:19706824; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25:1510-27; PMID:21764854; http://dx.doi.org/ 10.1101/gad.2051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25:795-800; PMID:21498569; http://dx.doi.org/ 10.1101/gad.2016211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460-70; PMID:21317241; http://dx.doi.org/ 10.1101/gad.2016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27:1447-61; PMID:23824538; http://dx.doi.org/ 10.1101/gad.219642.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100:15077-82; PMID:14657337; http://dx.doi.org/ 10.1073/pnas.2436255100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809-20; PMID:14638851; http://dx.doi.org/ 10.1172/JCI20039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621-35; PMID:17606641; http://dx.doi.org/ 10.1101/gad.1565707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367-81; PMID:17510285; http://dx.doi.org/ 10.1101/gad.1545107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon HU. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med 2013; 5:202ra123; PMID:24027027; http://dx.doi.org/ 10.1126/scitranslmed.3005864 [DOI] [PubMed] [Google Scholar]
- 73.Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res 2014; 12:485-90; PMID:24478461; http://dx.doi.org/ 10.1158/1541-7786.MCR-13-0614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun 2014; 5:3056; PMID:24445999; http://dx.doi.org/ 10.1038/ncomms4056 [DOI] [PubMed] [Google Scholar]
- 75.Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013; 504:296-300; PMID:24305049; http://dx.doi.org/ 10.1038/nature12865 [DOI] [PubMed] [Google Scholar]
- 76.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 2014; 4:905-13; PMID:24875860; http://dx.doi.org/ 10.1158/2159-8290.CD-14-0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pérez E, Das G, Bergmann A, Baehrecke EH. Autophagy regulates tissue overgrowth in a context-dependent manner. Oncogene 2014:in press; PMID:25174403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Granville CA, Warfel N, Tsurutani J, Hollander MC, Robertson M, Fox SD, Veenstra TD, Issaq HJ, Linnoila RI, Dennis PA. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and phenotypic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res 2007; 13:2281-9; PMID:17404113; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-2570 [DOI] [PubMed] [Google Scholar]
- 79.Seront E, Boidot R, Bouzin C, Karroum O, Jordan BF, Gallez B, Machiels JP, Feron O. Tumour hypoxia determines the potential of combining mTOR and autophagy inhibitors to treat mammary tumours. Br J Cancer 2013; 109:2597-606; PMID:24157830; http://dx.doi.org/ 10.1038/bjc.2013.644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koehl GE, Spitzner M, Ousingsawat J, Schreiber R, Geissler EK, Kunzelmann K. Rapamycin inhibits oncogenic intestinal ion channels and neoplasia in APC(Min/+) mice. Oncogene 2010; 29:1553-60; PMID:19966863; http://dx.doi.org/ 10.1038/onc.2009.435 [DOI] [PubMed] [Google Scholar]
- 81.Nam HY, Han MW, Chang HW, Lee YS, Lee M, Lee HJ, Lee BW, Lee HJ, Lee KE, Jung MK, et al. Radioresistant cancer cells can be conditioned to enter senescence by mTOR inhibition. Cancer Res 2013; 73:4267-77; PMID:23722550; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-3516 [DOI] [PubMed] [Google Scholar]
- 82.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533-41; PMID:21878654; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev Res 2010; 3:1066-76; PMID:20810672; http://dx.doi.org/ 10.1158/1940-6207.CAPR-10-0055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, Glicksman R, Chaib S, Zamiara P, Milosevic M, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res 2013; 19:6741-50; PMID:24141625; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-1787 [DOI] [PubMed] [Google Scholar]
- 85.Liu H, Scholz C, Zang C, Schefe JH, Habbel P, Regierer AC, Schulz CO, Possinger K, Eucker J. Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer Res 2012; 32:1627-37; PMID:22593441 [PubMed] [Google Scholar]
- 86.Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman NK, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov 2014; 4:773-80; PMID:24823863; http://dx.doi.org/ 10.1158/2159-8290.CD-14-0049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:21436444; http://dx.doi.org/ 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 88.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334:1573-7; PMID:22174255; http://dx.doi.org/ 10.1126/science.1208347 [DOI] [PubMed] [Google Scholar]