

Abstract
Cancer stem cells and their relatively differentiated progenitors coexist in dynamic equilibrium and are subject to bidirectional conversion. We recently showed that reprogramming transcription factors induce glioblastoma cells to become stem-like and tumor propagating via a mechanism involving changes in global DNA methylation and downregulation of miRNAs.
Keywords: cancer stem cell, DNA methylation, miRNAs, reprogramming transcription factors, tumorigenesis
In the early 1990s, characterization of well-defined cell surface markers led to the identification of undifferentiated precursor cells and distinct hematopoietic lineages derived from hematopoietic stem cells.1 Taking advantage of this groundbreaking development, Lapidot et al. demonstrated that CD34+/CD38− cell subsets were uniquely able to reconstitute acute myeloid leukemia in immunocompromised mice and rejuvenated interest in the cancer stem cell (CSC) hypothesis.2 Cell subpopulations similarly endowed with the capacities to efficiently propagate tumor xenografts with high histopathologic fidelity, self-renew, and generate transit amplifying progenitor cells with limited self-renewing capacity through asymmetric division were subsequently identified in breast carcinoma, glioblastoma, and other cancer types, thus extending the cancer stem cell hypothesis to solid 3
The origin of cancer stem cells (CSCs) in solid tumors remains unclear. The classic view originating from normal stem cell biology envisions the CSC as a distinct cellular entity at the pinnacle of a tumor cell hierarchy, with asymmetric division serving to maintain the CSC pool and generate bulk populations of more differentiated tumor progenitor cells with limited tumor-propagating capacities. Considerable experimental evidence from multiple laboratories and malignancies now shows that cells displaying the core criteria of CSCs (i.e., self-renewing, tumor propagating, multipotent) can arise through the dedifferentiation of non-tumor propagating tumor cell subsets, forcing a significant revision of the original strictly hierarchical model. Current models incorporate the inherent plasticity of malignant cells and their capacity to dynamically and bi-directionally alter phenotype and tumor-propagating capacity (e.g., “stemness”). Autocrine and microenvironment-derived molecular cues activate differentiating and dedifferentiating cell signaling cascades, transcriptional networks, and epigenetic mechanisms that regulate CSC pools and hence therapeutic resistance and tumor recurrence. Understanding these mechanisms is critical to developing more effective cancer therapeutics.
The discovery that a defined set of reprogramming transcription factors is sufficient to induce pluripotency and reprogram mature fibroblasts to an undifferentiated, stem-like state constituted a paradigm shift in the field of stem cell biology and greatly affected the way we look at the CSC.4 Since this discovery, expression of these same core reprogramming transcription factors has been found to correlate with cancer malignancy and poor patient prognosis. The finding that the gene expression profiles of embryonic stem cells (ESCs) closely resemble those of high-grade tumors suggested that similar transcriptional mechanisms drive de-differentiated phenotypes in non-malignant and malignant cells.5 Applying these discoveries of how non-malignant cells can be induced to dedifferentiate to the setting of malignancy has filled in missing mechanisms and added extra levels of complexity to the origins and nature of the CSC.
We now understand that the phenotypic heterogeneity of cancer cells in solid malignancies is supported by the ability of tumor cells, primed by the oncogenic genomic background, to dynamically transition between more differentiated and less differentiated states via mechanisms akin to induced plutipotency (i.e., spontaneous induced multipotency) and cellular reprogramming. Relevant findings in our laboratory demonstrated that oncogenic receptor tyrosine kinase signaling (i.e., hepatocyte growth factor (HGF)/c-MET signaling) drives the glioma stem cell phenotype by inducing reprogramming transcription factors and that inhibiting this axis in vivo depletes tumors of their stem-like tumor propagating cells.6,7 We also found that co-expression of the reprogramming transcription factors POU class 5 homeobox 1 (POUF1, best known as OCT4) and sex determining region Y-box 2 (SOX2) in tumor cells that lack stemness and tumor propagating potential induces de-differentiation and, more significantly, tumor propagating capacity. These reprogramming events were found to involve the direct activation of DNA methyl-transferase gene transcription, increased DNA methylation, and methylation-dependent repression of miRNA networks containing miRNAs that inhibit tumor cell stemness and tumor propagating potential.8
These newfound concepts and mechanisms of cancer cell plasticity have led to a re-examination of factors that might influence tumor propagating potential and tumor recurrence and new thought-provoking paradigms. One such concept relates to the role of tumor microenvironments and the concept of “stress-induced reprogramming” whereby unfavorable conditions, such as the hypoxic tumor microenvironment, inflammatory microenvironment, or stress induced by radiation/chemotherapy, activate reprogramming cascades that result in the de-differentiation of tumor cells to a more stem-like state with the ability to maintain or reconstitute the malignancy. As examples, Heddleston et al. found that hypoxia promotes self-renewal of glioma stem cells and non-stem cells through a process regulated by hypoxia-inducible factor 2α (HIF2α), OCT4, SOX2, NANOG, and v-myc avian myelocytomatosis viral oncogene homolog (c-MYC).9 Interestingly, Legadec et al. found that radiation therapy can cause fully differentiated breast cancer cells to de-differentiate into induced breast cancer stem cells.10 These findings establish the involvement of dynamically regulated de-differentiation mechanisms in CSC generation and maintenance and convey the possibility that reprogramming can be a side effect of therapeutic intervention that might contribute to treatment resistance and tumor recurrence. Recent advances in understanding the molecular drivers of the tumor-propagating CSCs open prospects for preventing these adverse consequences of current therapies. The discovery of potent CSC-inhibiting miRNAs opens new and exciting possibilities. Recent advances in nanomedicine now allow us to deliver nucleic acids effectively and with high selectivity in vitro and offer the potential for near-term in vivo application. We envision that combining current advances in nanomedicine with our growing understanding of CSC-inhibitory miRNAs will influence the way that we target tumor-propagating CSCs.
We now realize that the CSC population is very dynamic and that cells can gain or lose the stem-like phenotype depending on autocrine and environmental cues, resulting in heterogeneous populations of tumor cells that can either expand in a limited fashion (i.e., transit amplifying cells) or actually propagate tumors (i.e., CSCs) (Fig. 1). The dynamic transitions between these phenotypes depend upon the molecular cross talk between epigenetic modifications, reprogramming transcription factors, and coding and non-coding RNAs. Understanding the regulatory interactions between these elements has become a new and important area of research. Identifying the components and circuitries that generate and maintain CSCs will allow us to design more rational therapies to target phenotypic states that drive tumor growth and recurrence.
Figure 1.
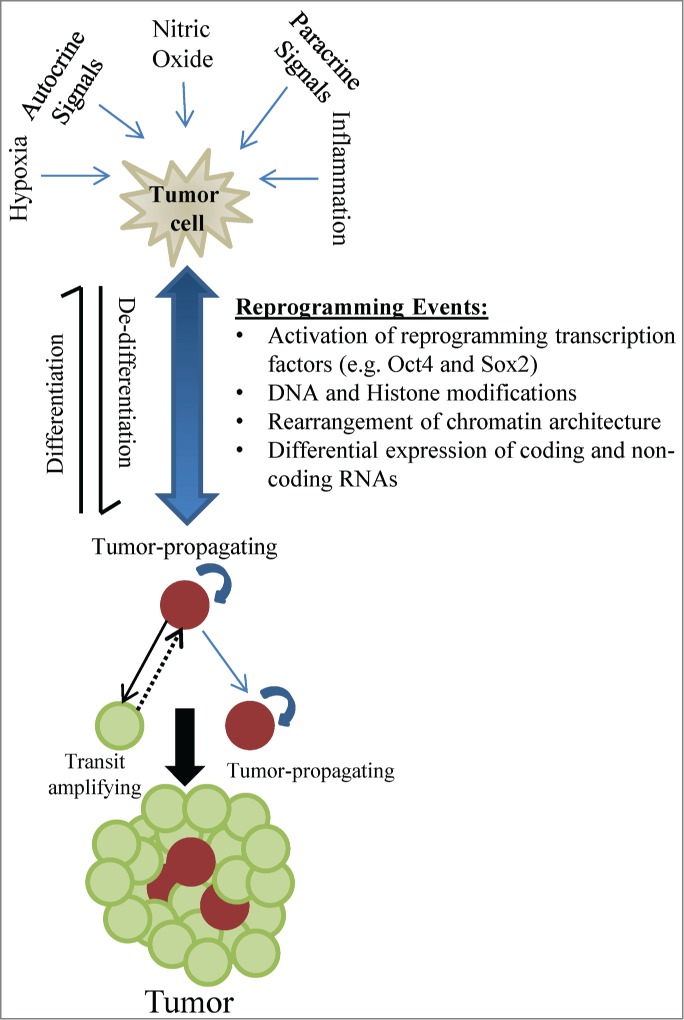
The CSC phenotype is dynamic and encompasses molecular changes that alter “stemness." This process is influenced by autocrine and paracrine pathways including environmental cues that modify the DNA methylation landscape and histone marks, regulate non-coding RNAs, and modulate the expression of transcription factors, all leading to fate-determining gene expression changes. De-differentiation and acquisition of stem cell qualities results in tumor cells with self-renewal capability, tumor-propagating capacity, and treatment resistance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH grants NS076759 (JL) and NS073611 (JL).
References
- 1.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science 1988; 241:58-62 ; PMID:2898810 [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine 1997; 3:730-37; PMID:9212098; http://dx.doi.org/ 10.1038/nm0797-730 [DOI] [PubMed] [Google Scholar]
- 3.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature; 2004; 432:396-401, PMID:15549107 http://dx.doi.org/ 10.1038/nature03128 [DOI] [PubMed] [Google Scholar]
- 4.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663-76; PMID:16904174 http://dx.doi.org/ 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res 2012; 72:576-80; PMID:22298594 http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rath P, Lal B, Ajala O, Li Y, Xia S, Kim J, Laterra J. In vivo c-Met pathway inhibition depletes human glioma xenografts of tumor-propagating stem-like cells. Transl Oncol 2013; 6:104-111; PMID:23556031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, Xia S, Trageser D, Guerrero-Cázares H, Eberhart CG, et al. . c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U S A 2011; 108:9951-9956; PMID:21628563; http://dx.doi.org/ 10.1073/pnas.1016912108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez-Bertoni H, Lal B, Li A, Caplan M, Guerrero-Cázares H, Eberhart CG, Quiñones-Hinojosa A, Glas M, Scheffler B, Laterra J, et al. . DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene 2014; PMID:25328136; http://dx.doi.org/ 10.1038/onc.2014.334 [ePub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heddleston JM, Li Z, McLendon RE, Hjelmeland AB. Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009; 8:3274-3284 ; PMID:19770585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012; 30:833-44; PMID:22489015; http://dx.doi.org/ 10.1002/stem.1058 [DOI] [PMC free article] [PubMed] [Google Scholar]