

Abstract
Autophagy and endoplasmic reticulum (ER) stress are involved in the development, progression, and chemoresistance of melanoma. We recently reported that oncogenic serine/threonine-protein kinase BRAF induces chronic ER stress, hence increasing baseline autophagy and promoting chemoresistance. The attenuation of ER stress restores basal autophagic activity and resensitizes melanoma cells to apoptosis.
Keywords: autophagy, BRAF, cancer, ER stress, melanoma
Abbreviations
- ASK1
apoptosis signal-regulating kinase 1
- ATF
activating transcription factor
- BRAF
serine/threonine-protein kinase B-raf
- BCL-2
B-cell lymphoma 2
- BCL-XL
BCL2-like 1 isoform 1
- BECN1
BECLIN 1
- eIF
eukaryotic translation initiation factor
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- GRP78
glucose-regulated protein 78kDa
- IRE1
inositol-requiring enzyme 1
- JNK
c-Jun N-terminal kinase
- MEK
mitogen-activated protein kinase kinase
- mTOR
mammalian target of rapamycin
- p38
mitogen-activated protein kinase 14
- PERK
double-stranded RNA activated protein kinase (PKR)-like endoplasmic reticulum kinase
- TBR3
mammalian homolog of Drosophila tribbles
- TRAF2
TNF receptor-associated factor 2
- UPR
unfolded protein response
- 4PBA
4-phenylbutyric acid
Proliferation and cell death are finely regulated and evolutionarily conserved physiologic processes that work in contraposition to play a pivotal role in the homeostasis of a multicellular organism. Indeed, in every organism several million cells are thought to die every second and this phenomenon of cellular demise is balanced by cell proliferation. This epic battle represents a fundamental process throughout the lifespan of an organism, since an unbalanced equilibrium between cell proliferation and death may result in tumor development.
Importantly, most tumors are characterized by an acquired desensitization to apoptotic stimuli, mainly due to dysfunctional apoptosis or deregulated proliferation, as a result of mutations, ablations, or dysfunction of key genes of the 2 signaling pathways.
Moreover, dysfunction or improper activation or inhibition of other molecular pathways may contribute to both tumor growth and unresponsiveness of cancer cells to therapy. Recent studies point to dysregulation of the unfolded protein response (UPR) and autophagy during tumor development/progression and/or under chemotherapeutic treatment that is associated with poor clinical outcome. Indeed, improper activity of these pathways negatively affects drug-induced apoptosis.1,2
Both endoplasmic reticulum (ER) stress and autophagy are primarily prosurvival pathways that are activated under stress conditions such as nutrient shortage and hypoxia, a typical feature of the solid tumor microenvironment, and under chemotherapy to actively counteract proapoptotic stimuli. Thus, although targeting ER stress and/or autophagy may provide a considerable benefit for cancer therapy, such potential is complicated by the interconnection of the 2 processes since each can regulate the other. Moreover, in this context the specific function of both ER stress and autophagy is strictly dependent on both tumor type and disease stage, as demonstrated in melanoma.3
Cutaneous metastatic melanoma is one of the most aggressive and difficult to treat forms of human malignancy, and moreover has an increasing incidence. Although mutation of the serine/threonine-protein kinase B-raf (BRAF), resulting in constitutive kinase activation, represents the most characteristic feature of this tumor (present in 70% of all melanomas), other mutations are also involved. Moreover, many reports indicate that dysregulation of autophagy and ER stress may represent 2 key signaling pathways involved in melanoma development, progression, and resistance to therapy, although an univocal interpretation is still far from evident.4,5 Moreover, the underlying genotype of an individual melanoma also appears to influence UPR and autophagic functions, with BRAF in particular appearing to have a direct effect on both pathways.6,7 Indeed, oncogenic BRAF reduces cell susceptibility to ER stress-induced apoptosis and concomitantly results in higher basal autophagic rates compared to BRAF wild type melanoma cells, although the underlying molecular mechanisms mediating this effect remain unclear.6,7 Recently, we and 2 other research groups independently identified 3 molecular pathways linking oncogenic BRAF activity and UPR induction in melanoma cells; these pathways involve (i) eukaryotic translation initiation factor 4E (eIF4E),8 (ii) mitogen-activated protein kinase 14 (MAPK14, also known as p38)9 or (iii) glucose-regulated protein 78kDa (GRP78)10. Inhibition of eIF4E phosphorylation as a result of oncogenic BRAF-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) activation results in protein hyperproduction and consequent UPR induction.8 Oncogenic BRAF also results in constitutive p38 activation, contributing to ER stress sensor activation and UPR tuning.9 Finally, mutated BRAF may also directly interact with GRP78, thus relieving its inhibitory activity on activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1), and double-stranded RNA activated protein kinase (PKR)-like endoplasmic reticulum kinase (PERK) and stimulating the UPR signaling pathway10 (Fig. 1).
Figure 1.
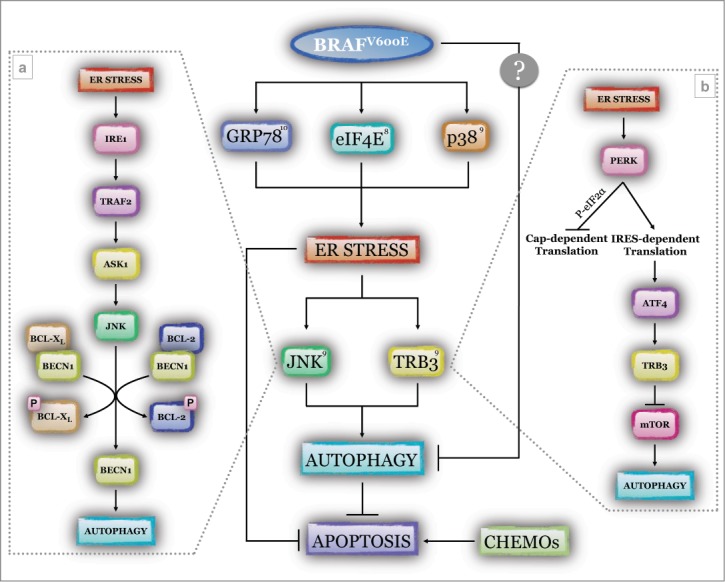
ER stress induced by oncogenic BRAF and modulation of basal autophagy in melanoma. Oncogenic serine/threonine-protein kinase B-raf carrying the V>E mutation (BRAFV600E) stimulates endoplasmic reticulum (ER) stress through the involvement of at least 3 major factors, glucose-regulated protein 78kDa (GRP78), eukaryotic translation initiation factor 4E (eIF4E), and mitogen-activated protein kinase 14 (p38). Through direct interaction with GRP78, mutant BRAF relieves its inhibitory activity toward the 3 stress sensors, activating transcription factor 6 (ATF6), inositol-requiring enzyme (IRE1), and double-stranded RNA activated protein kinase (PKR)-like endoplasmic reticulum kinase (PERK),10 and thus inducing the unfolded protein response (UPR). Stimulation of the activity of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) signaling pathway drives phosphorylation of the translational factor eIF4E, hyperstimulating the translational signaling pathway and resulting in protein accumulation in the ER compartment;8 activation of p38 (through protein phosphorylation) drives the activation of the above mentioned ER stress sensors, resulting in UPR.9 ER stress in turn enhances the activity of the autophagic process through the PERK/activating transcription factor 4 (ATF4)/mammalian homolog of Drosophila tribbles (TRB3) and the IRE1/TNF receptor-associated factor 2 (TRAF2)/apoptosis signal-regulating kinase 1 (ASK1)/c-Jun N-terminal kinase (JNK) axes. In the first signaling pathway (B), active PERK phosphorylates the eukaryotic translation initiation factor 2α (eIF2α), a factor controlling the initiation of cap-dependent protein translation, thus stimulating the IRES-dependent translational process. This favors accumulation of the transcription factor ATF4 that in turn stimulates the synthesis of TRB3, which induces the autophagic process through inhibition of mammalian target of rapamycin (mTOR) activity. In parallel (A), active IRE1 recruits and activates the cytosolic kinase ASK1 through the adapter molecule TRAF2, which in turn activates JNK. The latter phosphorylates the BECLIN 1 (BECN1) inhibitory partners B-cell lymphoma 2 (BCL-2) and BCL2-like 1 isoform 1 (BCL-XL), resulting in BECN1 release and subsequent autophagy induction. Therefore, mutant BRAF-mediated dysregulation of prosurvival pathways such as ER stress and autophagy sustains tumor development and progression, and desensitizes cancer cells to therapy-induced apoptosis (CHEMOs).
ER stress activation per se enhances tumor growth and inhibits cell death induction,1 thus promoting tumor progression and chemoresistance. Moreover, oncogenic BRAF-induced UPR also results in autophagy enhancement in melanoma cells, contributing to desensitization of cells to apoptosis induction.9,10 Importantly, we have revealed the underlying molecular mechanisms linking the 2 pathways in BRAF mutant melanoma cells based on activation of (i) the PERK/activating transcription factor 4 (ATF4) axis of UPR, resulting in upregulation of mammalian homolog of Drosophila tribbles (TRB3) and subsequent inhibition of mammalian target of rapamycin (mTOR) and consequent autophagy induction; ii) the IRE1/TNF receptor-associated factor 2/apoptosis signal-regulating kinase 1/c-Jun N-terminal kinase (IRE1/TRAF2/ASK1/JNK) axis, thus resulting in phosphorylation of B-cell lymphoma 2 (BCL-2)/BCL2-like 1 isoform 1 (BCL-XL), BECLIN 1 (BECN1) release, and autophagy induction9 (Fig. 1).
Inhibition of BRAF-mediated UPR using the chemical chaperone 4-phenylbutyric acid (4PBA) clearly resulted in basal autophagy inhibition and, crucially, resensitization to chemotherapy-stimulated melanoma cell death.9
Collectively, these data indicate that, although autophagy dysregulation undoubtedly has a major impact in melanoma, oncogenic BRAF-stimulated UPR assumes a pivotal role in melanoma tumor progression and acquired chemoresistance and, furthermore, directly controls the autophagic process.
In conclusion, targeted therapies that attenuate ER stress may represent a novel and effective therapeutic strategy for melanoma with oncogenic BRAF.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Funding
This work was supported by grants from AIRC (MFAG-11743 to MC, IG2012 to GMF and IG2010 to MP), the Italian Ministry of University and Research (PRIN 2012 and FIRB Accordi di Programma 2011), the Italian Ministry of Health (Ricerca Finalizzata and Ricerca Corrente), the British Skin Foundation, The JGW Patterson Foundation, and The Newcastle Healthcare Charity, UK.
References
- 1.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer 2014; 14:581–97; PMID:25145482; http://dx.doi.org/ 10.1038/nrc3800 [DOI] [PubMed] [Google Scholar]
- 2.Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol 2014; pii: S0962-8924(14)00158-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corazzari M. ER stress & autophagy in cancer: contenders or partners in crime? Int J Mol Biol Biochem 2013; 1:23–28 [Google Scholar]
- 4.Corazzari M, Fimia GM, Lovat PE, Piacentini M. Why ia autophagy important for Melanoma - molecular mechanisms and therapeutic implications. Semin Cancer Biol 2013; 23:337–43; PMID:23856558; http://dx.doi.org/ 10.1016/j.semcancer.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 5.Hersey P, Zhang XD. Adaptation to ER stress as a driver of malignancy and resistance to therapy in human melanoma. Pigment Cell Melanoma Res 2008; 21:358–67; PMID:18476909; http://dx.doi.org/ 10.1111/j.1755-148X.2008.00467.x [DOI] [PubMed] [Google Scholar]
- 6.Armstrong JL, Corazzari M, Martin S, Pagliarini V, Falasca L, Hill DS, Ellis N, Al Sabah S, Redfern CPF, Fimia GM, et al. . Oncogenic B-RAF Signaling in Melanoma Impairs the Therapeutic Advantage of Autophagy Inhibition. Clin Cancer Res 2011; 17:2216–26; PMID:21270111; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-3003 [DOI] [PubMed] [Google Scholar]
- 7.Corazzari M, Lovat PE, Armstrong JL, Fimia GM, Hill DS, Birch-Machin M, Redfern CP, Piacentini M. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: The role of stress proteins ERdj5 and ERp57. Br J Cancer 2007; 96: 1062–1071; PMID:17353921; http://dx.doi.org/ 10.1038/sj.bjc.6603672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croft A, Tay KH, Boyd SC, Guo ST, Jing CC, Lai F, Tseng HY, Jin L, Rizos H, Hersey P, et al. . Oncogenic activation of MEK/ERK primes melanoma cells for adaptation to endoplasmic reticulum stress. J Invest Dermatol 2014; 134:488–97; PMID:23921951; http://dx.doi.org/ 10.1038/jid.2013.325 [DOI] [PubMed] [Google Scholar]
- 9.Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ 2014; PMID:25361077; http://dx.doi.org/ 10.1038/cdd.2014.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma HX, Piao SF, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levis S, Hu J, Zhang G, Lazova R, et al. . Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest 2014; 124:1406–17; PMID:24569374; http://dx.doi.org/ 10.1172/JCI70454 [DOI] [PMC free article] [PubMed] [Google Scholar]