

Abstract
DNA double-strand breaks (DSBs) are highly deleterious lesions and their misrepair can promote genomic instability, a hallmark of cancer. DNA-end resection is a cell cycle-regulated mechanism that is required for the faithful repair of DSBs. We recently discovered that the anaphase-promoting complex/cyclosome-Cdh1 (APC/CCdh1) ubiquitin ligase is responsible for the timely degradation of CtBP-interacting protein (CtIP), a key DNA-end resection factor, providing a new layer of regulation of DSB repair in human cells.
Keywords: anaphase promoting complex/cyclosome (APC/C), cell cycle, Cdh1, CtIP, DNA damage, DNA double-strand break repair, DNA-end resection, genomic instability, ubiquitin-dependent proteolysis
Genomic instability is a hallmark of cancer cells and predominantly arises as a result of defects in components of the DNA damage response (DDR) machinery.1 DNA double-strand breaks (DSBs) are among the most dangerous lesions a cell can encounter as they could result in cancer-promoting chromosomal rearrangements. To optimally deal with DSBs, cells have evolved a number of repair pathways including non-homologous end joining (NHEJ) and homologous recombination (HR).1 NHEJ is fast and operates throughout the cell cycle, whereas HR is slower and the predominant repair mechanism during S phase. However, only HR is able to faithfully restore the original information by using the undamaged sister chromatid as a template for repair. HR is initiated by DNA-end resection, which requires the collaborative action of several enzymes, most importantly CtBP-interacting protein (CtIP, also known as RBBP8) and the MRE11-RAD50-NBS1 complex.2 Moreover, DNA-end resection is considered a critical step in selection of the DSB repair pathway, as the generation of single-stranded DNA (ssDNA) licenses HR and impairs NHEJ. Consequently, mechanisms regulating DNA-end resection are considered pivotal for the maintenance of genome stability.
The anaphase-promoting complex/cyclosome (APC/C), a multisubunit E3 ubiquitin ligase, orchestrates cell cycle progression by targeting critical cell cycle regulator proteins for proteasomal degradation.3 APC/C activity is mainly controlled through the mutually exclusive binding of 2 coactivators, cell division cycle protein 20 (Cdc20) and Cdc20-homolog 1 (Cdh1). APC/CCdc20 predominantly targets destruction box (D box)-containing substrates during the metaphase to anaphase transition. In anaphase, Cdc20 is replaced by Cdh1, which mostly recognizes KEN box-containing substrates and keeps APC/C active until the end of G1 phase. Although APC/CCdh1 is normally kept inactive during S and G2 phase, premature activation has been reported after genotoxic stress in G2 to establish an efficient DNA damage checkpoint.4,5 Moreover, loss of Cdh1 was shown to result in increased genomic instability.6 Consistent with data from the Malumbres laboratory, we found that Cdh1 downregulation leads to hypersensitivity to DSB-inducing agents.7,8 This prompted us to investigate whether APC/CCdh1 plays a more prominent role in DSB repair.
Indeed, using an integrated proteomics and bioinformatics approach, we identified the DNA-end resection factor CtIP as a novel APC/CCdh1 substrate.8 Protein sequence alignments highlighted the presence of an evolutionary conserved KEN box in CtIP, mutation of which resulted in defective Cdh1 interaction and CtIP ubiquitination. Furthermore, we showed that APC/CCdh1 exerts a dual control over CtIP, triggering its proteasome-dependent degradation both during G1 and following DSB formation in G2. First, we found that interfering with APC/C activity following mitotic exit led to increased CtIP protein levels in G1 cells. Second, in G2 phase following exposure to ionizing irradiation (IR), we showed that the CtIP KEN box mutant displays increased protein stability and prolonged retention at sites of DSBs when compared with the wild-type CtIP protein. As a logical consequence, cells expressing this mutant exhibit increased hyperphosphorylation of the replication protein A 32-kDa subunit (RPA2), a widely accepted marker for DNA-end resection.2 Although technically challenging, it would be interesting to further investigate whether increased ssDNA formation was due to either a fixed number of DSBs undergoing more extensive resection or a greater number of DSBs undergoing normal resection. Indeed, it has been estimated that during G2 phase HR is responsible for the repair of 20–30% of IR-induced DSBs, whereas the majority of lesions are addressed by NHEJ.9 Thus, one may speculate that in a situation where CtIP cannot be degraded after having successfully initiated the resection of a specific subset of DSBs, it acquires the potential to process DSBs that are otherwise preferentially addressed by NHEJ. We therefore hypothesized that under conditions of high genotoxic stress (e.g., after radiotherapy), cells prematurely activate APC/CCdh1 and degrade CtIP in an attempt to minimize DNA-end resection, thereby increasing the capacity of NHEJ.
Based on our data, we concluded that APC/CCdh1-mediated degradation of CtIP contributes to the regulation of DNA-end resection and therefore plays an unexpected role in the coordination of DSB repair pathways (Fig. 1). In G1 cells, when the sister chromatid template is unavailable for HR-mediated DSB repair, APC/CCdh1 targets CtIP to minimize DNA-end resection and allow efficient NHEJ to maintain genome stability. This scenario is broadly consistent with recent findings from the Löbrich laboratory showing that CtIP-dependent resection of DSBs in G1 enhances the formation of chromosomal translocations.10 In contrast, because of the existing crosstalk and fine balance between HR and NHEJ, the role of APC/CCdh1 in the regulation of DSB repair in G2 cells is not that straightforward and most likely involves the targeting of other DNA damage response factors besides CtIP. In fact, we found that the hyper-resection phenotype observed in cells expressing the CtIP KEN box mutant does not correlate with increased RAD51-dependent HR, indicating that the magnitude of DNA-end resection has to be tightly regulated in order to allow successful completion of HR. Furthermore, our proteomic approach identified other DDR factors as candidate substrates of the APC/C (e.g., RIF1, SMC5, and MDC1). A detailed characterization of additional APC/CCdh1 substrates involved in the regulation of DSB repair, in addition to CtIP, may eventually provide us with a more comprehensive picture of how APC/CCdh1 contributes to the maintenance of genome stability.
Figure 1.
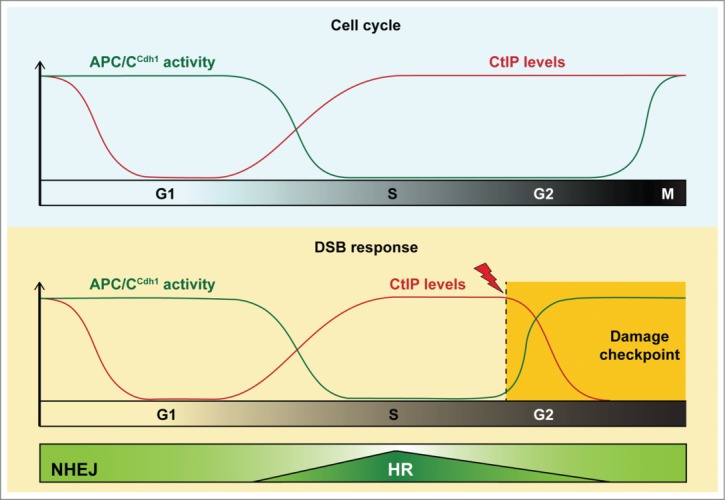
The ubiquitin ligase APC/CCdh1 controls cell cycle-dependent repair of DNA double-strand breaks (DSBs) through the degradation of CtIP. During normal cell cycle progression (upper panel), anaphase-promoting complex/cyclosome-Cdh1 (APC/CCdh1) targets CtIP for ubiquitin-dependent proteolysis exclusively in G1. At the G1/S transition, APC/CCdh1 activity decreases, allowing CtIP protein levels to increase. Similarly, in the case of DNA DSB formation in G2 cells (lower panel), APC/CCdh1 becomes prematurely activated, resulting in accelerated CtIP protein turnover and decreased DNA-end resection. We propose that this regulatory circuit contributes to the regulation of DNA-end resection and thus the balance between non-homologous end joining (NHEJ, predominant in G1 and G2 phase) and homologous recombination (HR, predominant in S phase).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/ 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature 2007; 450:509-14; PMID:17965729; http://dx.doi.org/ 10.1038/nature06337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pines J. Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol 2011; 12:427-38; PMID:21633387; http://dx.doi.org/ 10.1038/nrm3132 [DOI] [PubMed] [Google Scholar]
- 4.Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, Saya H. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J 2001; 20:6499-508; PMID:11707420; http://dx.doi.org/ 10.1093/emboj/20.22.6499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008; 134:256-67; PMID:18662541; http://dx.doi.org/ 10.1016/j.cell.2008.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcí-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, Malumbres M. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol 2008; 10:802-11; PMID:Can't; http://dx.doi.org/ 10.1038/ncb1742 [DOI] [PubMed] [Google Scholar]
- 7.Eguren M, Álvarez-Fernández M, García F, López-Contreras AJ, Fujimitsu K, Yaguchi H, Luque-García JL, Fernandez-Capetillo O, Muñoz J, Yamano H, et al.. A Synthetic Lethal Interaction between APC/C and Topoisomerase Poisons Uncovered by Proteomic Screens. Cell Rep 2014; 6:670-83; PMID:24508461; http://dx.doi.org/ 10.1016/j.celrep.2014.01.017 [DOI] [PubMed] [Google Scholar]
- 8.Lafranchi L, de Boer HR, de Vries EG, Ong S-E, Sartori AA, van Vugt MA. APC/CCdh1 controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J 2014; 33:2860-79; PMID:25349192; http://dx.doi.org/ 10.15252/embj.201489017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, et al.. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 2011; 30:1079-92; PMID:21317870; http://dx.doi.org/ 10.1038/emboj.2011.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barton O, Naumann SC, Diemer-Biehs R, Künzel J, Steinlage M, Conrad S, Makharashvili N, Wang J, Feng L, Lopez BS, et al.. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J Cell Biol 2014; 206:877-94; PMID:25267294; http://dx.doi.org/ 10.1083/jcb.201401146 [DOI] [PMC free article] [PubMed] [Google Scholar]