

Abstract
Inflammation and mutation of the tumor suppressor p53 are two apparently unrelated conditions that are strongly associated with cancer initiation and progression. We recently reported that gain-of-function mutant p53 modifies the response of cancer cells to inflammatory signals by binding a cytoplasmic tumor suppressor protein involved in TNFα signaling.
Keywords: DAB2IP, AIP1, tumor necrosis factor, cancer cell invasivity, basal-like breast cancer
The tumor suppressor gene TP53 is frequently mutated in cancer, and missense p53 mutants (mutp53) are stable proteins that may acquire novel potentially oncogenic activities (gain-of-function or GOF mutants).1
Various lines of evidence link p53 mutation to proinflammatory behavior of cancer cells. In particular, mutp53 can enhance nuclear factor kappa-B (NF-κB) activity in response to the proinflammatory cytokine tumor necrosis factor α (TNFα), and a correlation between nuclear p65 NF-κB (RelA) staining and mutant p53 can be observed in cancer samples.2 Most notably, a recent study demonstrated that germline p53 mutation dramatically accelerates colon cancer in a mouse model of colitis.3 The latter work provided evidence that mutp53 sustains NF-κB activity in conditions of chronic TNFα stimulation, and concluded that p53 mutation predisposes to colitis-related cancer by enforcing and maintaining a proinflammatory microenvironment that favors cell transformation. Conceptually, this conclusion may be extended to other chronic inflammatory conditions.
Considering that mutation of p53 often occurs at later stages during tumorigenesis, we speculated that p53 mutations might also affect inflammation-dependent progression and aggressiveness of already initiated cancers. Indeed, we observed that metastatic cancer cell lines with various p53 point mutations respond to an inflammatory milieu by increasing their invasive behavior.4 We confirmed that mutp53 amplifies TNF-induced expression of NF-κB target genes, but we also noticed that mutp53 dampens expression of TNF-inducible genes that depend on the ASK1 (apoptosis signal-regulating kinase)/JNK (c-Jun N-terminal kinase)/AP-1 (transcription factor AP-1) axis.
Previous studies reported that mutp53 binds to the promoters of inflammatory genes.2,3 We found that mutp53 also acts at another level, directly downstream of the TNF receptor, by physically interacting with the tumor suppressor disabled homolog-2 interacting protein (DAB2IP).
DAB2IP (also known as ASK1 interacting protein [AIP1]) is a cytoplasmic Ras-GTPase activating protein that also functions as a signaling scaffold to modulate the cell's response to multiple oncogenic extracellular signals, including TNFα.5,6 We observed that mutp53 binds DAB2IP in the cytoplasm, interfering with its physiological interactions; this reduces TNF-induced activation of the ASK1/JNK axis, thereby promoting activation and nuclear translocation of NF-κB (Fig. 1).
Figure 1.
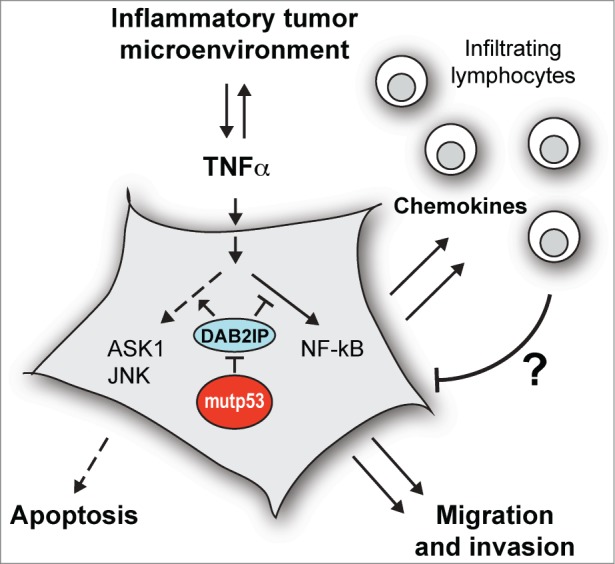
Mutant p53 binds the signal transducer DAB2IP in the cytoplasm and interferes with its functions. This favors activation of the NF-kB transcription factor and reduces activation of the ASK1/JNK kinases, resulting in a gene expression program that stimulates cell invasion and protects from cell death, thus increasing cancer aggressiveness. This same program includes expression of powerful chemokines that can recruit immune cells to the tumor, potentially improving response to therapy. ASK1, apoptosis signal regulating kinase-1; DAB2IP, disabled homolog-2 interacting protein; JNK, c-jun N-terminal kinase; mutp53, missense mutant p53; NF-κB, nuclear factor kappa-B; TNFα, tumor necrosis factor-α.
To conclusively define such nuclear-independent mutp53 activity, we demonstrated that a cytoplasmic mutant p53 (lacking nuclear localization signals) is sufficient to mediate a proaggressive response to inflammatory cytokines in cancer cells. This is a paradigmatic example of cytoplasmic gain-of-function.
The idea that mutant p53 can have cytoplasmic functions is plausible, but to date has suffered from the lack of strong experimental evidence; we think that our data now provide convincing support for this concept. Further support for cytoplasmic GOF is provided by the recent demonstration that mutp53 can bind the α subunit of AMP-activated protein kinase (AMPK) in cancer cells, thus affecting its functions.7 Mutant p53 is found in variable but significant amounts in the cytoplasm of cancer cells, and it is likely that altered cellular localization—in addition to altered conformation—leads to the formation of novel protein complexes.
Interestingly, p53 nonsense or frameshift mutations at positions immediately before the nuclear localization signals are found in human cancers (UMD TP53 mutation database, http://www.p53.fr). Such mutants are predicted to encode monomeric p53 proteins with normal conformation of the DNA-binding domain, but a predominantly cytoplasmic localization. We previously reported that DAB2IP can interact with wild-type p53,8 therefore such mutants could bind DAB2IP in the cytoplasm and may have a specific oncogenic GOF in an inflammatory context. This is an intriguing hypothesis that we are currently exploring.
Another stimulating observation emerged from in silico studies. Survival analysis of breast cancer patients suggested that tumors with mutant p53 are associated with a better outcome if they express a group of genes (signature) that we have linked to the TNF/mutp53 axis.4 At first glance this observation seems paradoxical, since the TNF/mutp53 axis promotes an aggressive phenotype in cultured cells and nude mice. We think this apparent contradiction reflects the influence of a parameter that is missing in most experimental models: the host immune system. In fact, our data suggest that mutp53-modulated activation of NF-κB endows cancer cells with proinvasive and prosurvival activities but, at the same time, triggers expression of chemokines that can recruit leukocytes to the tumor—a phenomenon that may favor tumor elimination.9,10 In other words, to unleash the tumorigenic forces of NF-κB mutp53 necessarily exposes cancer cells to the immune system, which could dramatically affect the prognosis.
This hypothesis may open a number of interesting perspectives. For instance, integrating analysis of p53 mutation with the immunoscore10 (i.e., quantity and type of infiltrating immune cells) in cancer biopsies may improve prediction of the clinical outcome in mammary tumors. Also, given the double-edged effect of TNFα on cancer cells, the mutation status of p53 may be used to help clinical choices in cancer therapy.
Clearly, our observations also raise a number of questions. According to our analyses, mutp53 cancers expressing low levels of chemokines linked to the TNF/mutp53 axis tend to have a negative prognosis. What happens in those cancers? Are these cells less dependent on the NF-κB activity for their survival/dissemination? Or have they evolved a mechanism to uncouple expression of oncogenic NF-κB target genes from immunostimulatory chemokines?
In vitro models and xenografts in immunocompromised mice have allowed us to explore the proinvasive and aggressive side of this molecular axis and uncover part of its mechanism. Now, the challenge is to better understand the potential impact of mutp53 in the complex interaction of cancer cells with the host immune system, and possibly develop such knowledge into strategies to improve therapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by funding from AIRC (Italian Association for Cancer Research, IG 14173), Università di Trieste (FRA 2012), and a postdoctoral fellowship from FIRC (Fondazione Italiana Ricerca sul Cancro).
References
- 1.Muller PAJ, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013; 15:2–8; PMID:23263379; http://dx.doi.org/ 10.1038/ncb2641 [DOI] [PubMed] [Google Scholar]
- 2.Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, Kalis M, Levrero M, Strano S, Gorgoulis VG, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res 2007; 67:2396-401; PMID:17363555; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2425 [DOI] [PubMed] [Google Scholar]
- 3.Cooks T, Pateras Ioannis S, Tarcic O, Solomon H, Schetter Aaron J, Wilder S, Lozano G, Pikarsky E, Forshew T, Rozenfeld N, et al. Mutant p53 Prolongs NF-kB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer Cell 2013; 23:634-46; PMID:23680148; http://dx.doi.org/ 10.1016/j.ccr.2013.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Di Minin G, Bellazzo A, Dal Ferro M, Chiaruttini G, Nuzzo S, Bicciato S, Piazza S, Rami D, Bulla R, Sommaggio R, et al. Mutant p53 Reprograms TNF Signaling in Cancer Cells through Interaction with the Tumor Suppressor DAB2IP. Mol Cell 2014; 56:617-29; PMID:25454946; http://dx.doi.org/ 10.1016/j.molcel.2014.10.013 [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Zhang R, Luo Y, D'Alessio A, Pober JS, Min W. AIP1/DAB2IP, a novel member of the Ras-GAP family, transduces TRAF2-induced ASK1-JNK activation. J Biol Chem 2004; 279:44955-65; PMID:15310755; http://dx.doi.org/ 10.1074/jbc.M407617200 [DOI] [PubMed] [Google Scholar]
- 6.Wu K, Liu J, Tseng SF, Gore C, Ning Z, Sharifi N, Fazli L, Gleave M, Kapur P, Xiao G, et al. The role of DAB2IP in androgen receptor activation during prostate cancer progression. Oncogene 2014; 33:1954-63; PMID:23604126; http://dx.doi.org/ 10.1038/onc.2013.143 [DOI] [PubMed] [Google Scholar]
- 7.Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell 2014; 54:960-74; PMID:24857548; http://dx.doi.org/ 10.1016/j.molcel.2014.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lunardi A, Di Minin G, Provero P, Dal Ferro M, Carotti M, Del Sal G, Collavin L. A genome-scale protein interaction profile of Drosophila p53 uncovers additional nodes of the human p53 network. Proc Natl Acad Sci U S A 2010; 107:6322-7; PMID:20308539; http://dx.doi.org/ 10.1073/pnas.1002447107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012; 12:298-306; PMID:22419253; http://dx.doi.org/ 10.1038/nrc3245 [DOI] [PubMed] [Google Scholar]
- 10.Galon J, Angell HK, Bedognetti D, Marincola FM. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity 2013; 39:11-26; PMID:23890060; http://dx.doi.org/ 10.1016/j.immuni.2013.07.008 [DOI] [PubMed] [Google Scholar]