

Abstract
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that is frequently mutated or overexpressed in a large number of tumors such as carcinomas or glioblastoma. Inhibitors of EGFR activation have been successfully established for the therapy of some cancers and are more and more frequently being used as first or later line therapies. Although the side effects induced by inhibitors of EGFR are less severe than those observed with classic cytotoxic chemotherapy and can usually be handled by out-patient care, they may still be a cause for dose reduction or discontinuation of treatment that can reduce the effectiveness of antitumor therapy. The mechanisms underlying these cutaneous side effects are only partly understood. Important questions, such as the reasons for the correlation between the intensity of the side effects and the efficiency of treatment with EGFR inhibitors, remain to be answered. Optimized adjuvant strategies to accompany anti-EGFR therapy need to be found for optimal therapeutic application and improved quality of life of patients. Here, we summarize current literature on the molecular and cellular mechanisms underlying the cutaneous side effects induced by EGFR inhibitors and provide evidence that keratinocytes are probably the optimal targets for adjuvant therapy aimed at alleviating skin toxicities.
Keywords: EGFR, cetuximab, erlotinib, rash, barrier defect, cancer therapy
Abbreviations
- ADAM
disintegrin and metalloproteinase
- AMP
antimicrobial peptide
- AREG
amphiregulin
- BD
betadefensin
- CRC
colorectal cancer
- DC
dendritic cell
- DETC
dendritic epidermal T-cells
- EGFR-I
EGFR inhibitor
- EREG
epiregulin
- GEMM
genetically modified mouse model
- GMCSF
granulocyte-macrophage colony-stimulating factor
- HBEGF
heparin-binding EGF
- KRAS
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- MyD88
myeloid differentiation primary response gene 88
- SALT
Skin associated lymphoid tissue
- TGM
transglutaminase
- TKI
tyrosine kinase inhibitor
- TRPV
transient receptor potential cation channel, subfamily V
- wa
waved
Introduction
Our understanding of the molecular mechanisms at the basis of cancer has substantially progressed over the past decades, to a large extent due to the study of genetically engineered mouse models (GEMMs). More recently, the advent of automated large-scale analysis of cancer patient material has boosted this field and paved the way for new avenues toward understanding this debilitating disease. It is, however, still a challenge to effectively translate results from basic research into clinical applications.
One of the first molecules to be targeted based on its mutation, high level of expression, and involvement in tumor cell proliferation and survival was the epidermal growth factor receptor (EGFR, also known as ErbB1). Numerous in vitro studies have evaluated the antiproliferative potential of different EGFR inhibitors (EGFR-I) such as anti-EGFR antibodies or tyrosine kinase inhibitors (TKIs),1,2 and inhibition of angiogenesis and metastasis has been shown using in vivo models.3,4 Although the promising results from preclinical studies did not entirely hold true in the clinic there is no doubt that anti-EGFR therapy results in a significant benefit for specific cancer patients when applied either alone or in combination with radiation therapy or chemotherapy. However, a large number of patients experience adverse events that, although usually moderate, in some cases necessitate dose reduction or termination of therapy. Additionally, in the course of therapy tumors may upregulate other tyrosine kinases to escape anti-EGFR therapy.5 Future therapeutic strategies will aim at targeting several tyrosine kinases simultaneously, with the disadvantage of potentially increased side effects. Therefore, understanding the mechanisms underlying the side effects and their management, and also how these side effects correlate with the efficacy of the therapy, will be important for improving the effectiveness of anti-EGFR therapy. This review will give an overview of current knowledge of the pathomechanisms underlying adverse events in the skin of EGFR-I–treated patients.
The Epidermal Growth Factor Receptor
The epidermal growth factor receptor (EGFR, also known as ErbB1) is a receptor tyrosine kinase of the ErbB family that additionally consists of ErbB2/neu, ErbB3, and ErbB4. Upon binding of EGFR-specific ligands such as epidermal growth factor (EGF), amphiregulin (AREG), transforming growth factor α (TGFα), epigen, or ligands shared with ErbB4, such as epiregulin (EREG), betacellulin, or heparin-binding epidermal growth factor (HB-EGF) a conformational change of the EGFR is induced that allows homo- or hetero-dimerization with other family members (Fig. 1A, B).6
Figure 1.
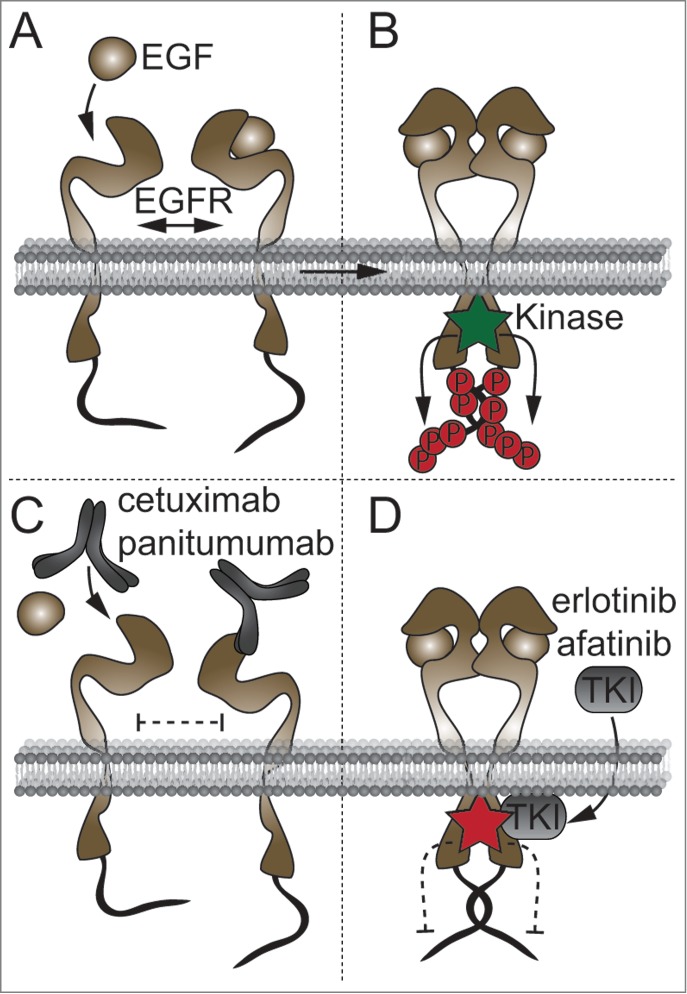
Principles of EGFR activation and inhibition. (A) In the absence of ligand, EGFR remains in a conformation that inhibits dimerization. (B) Upon ligand binding, the resultant structural change allows homo- or hetero-dimerization with members of the ErbB family, resulting in autophosphorylation of the intracellular tyrosine kinase domain. Kinase activity induces phosphorylation of tyrosines at the C-terminal tail, inducing downstream signaling. (C, D) Therapeutic anti-EGFR antibodies bind the extracellular domain of EGFR and inhibit ligand binding (C), whereas tyrosine kinase inhibitors compete for ATP binding at the tyrosine kinase domain, thereby inhibiting kinase activity (D).
EGFR ligands are generated as membrane-bound pro-forms that require cleavage by proteases to induce autocrine and paracrine EGFR signaling. Ectodomain shedding of EGFR ligands is mainly performed by a disintegrin and metalloproteinase (ADAM) proteins 10 and 17.7 However, juxtacrine signaling by membrane-bound EGFR ligands has also been reported and it is not yet clear whether these different modes of action have distinct biological consequences.8 Dependent on ligand and dimerization partners, EFGR activation may result in signaling via MAPK, STATs, PI3K, or PLCγ.9 Analysis of mice lacking EGFR revealed that EGFR plays an essential role during fetal development and also in tissue homeostasis during adult life.10-14 Mutant mice develop neurodegeneration shortly after birth and display defects in several epithelial compartments depending on the genetic background.10,13-15 The skin is particularly affected in EGFR-deficient mice, showing impaired hair follicle development and hair growth and strong inflammation.16-18 Recently, a child carrying an inherited loss-of-function mutation of the EGFR was reported who showed lifelong inflammation in the skin, gut, and lung that caused early death of the infant, highlighting the importance of EGFR signaling for establishment and maintenance of tissue homeostasis.19
EGFR Inhibitors
Overexpression of EGFR or its ligands and activating mutations in the EGFR signaling pathway may lead to epithelial neoplasms and can be found in a large number of cancers in various tissues.20-22 EGFR activation promotes multiple tumorigenic processes by regulating proliferation, cell survival, angiogenesis, and metastasis.23 Knowing this, strategies aimed at inhibiting EGFR signaling by targeted therapies were developed. Currently, 2 strategies to inhibit EGFR signaling—monoclonal antibodies and tyrosine kinase inhibitors (TKI)—have been approved for the treatment of cancer either alone or in combination with cytotoxic therapies such as standard chemotherapy or radiation therapy. Starting in the 1980s, several monoclonal antibodies against the ligand binding domain of EGFR have been developed.24 Anti-EGFR antibodies currently used in the clinic are cetuximab and panitumumab. These agents are highly specific for EGFR and, by blocking ligand binding, prevent the conformational change in EGFR necessary for dimerization (Fig. 1C).25 Cetuximab and panitumumab are approved for the treatment of patients with squamous cell carcinoma and colorectal cancer (CRC). However, the efficacy of cetuximab against CRC is restricted to defined patient collectives expressing characteristic biomarker patterns. Mutation at codon 12 of KRAS is negatively predictive for the response to cetuximab whereas wild-type (wt) KRAS or the KRAS G13D mutation has no prognostic effect.26,27 Not all patients with wt KRAS respond to cetuximab or panitumumab; other prognostic parameters in patients with wt KRAS codon12 include BRAF mutation, PI3K mutation, localization in the left colon (reduced likelihood of KRAS or BRAF mutation), and AREG expression in the tumor tissue, whereas EREG expression is a positive prognostic factor for the response to cetuximab in metastatic CRC that may be independent of the KRAS mutation status.28-31 Additionally, AREG polymorphisms have been shown to affect the efficiency of cetuximab in CRC.32
As an alternative strategy to inhibit EGFR, signaling inhibitors of the tyrosine kinase domain have been developed (Fig. 1D). These small molecules bind to the intracellular tyrosine kinase domain and inhibit receptor autophosphorylation by competing with ATP. TKIs have varying affinities for the ATP binding sites of other tyrosine kinases, for example ErbB2 and VEGR, and are therefore not as specific for EGFR as anti-EGFR antibodies. However, TKIs like erlotinib, gefitinib, or afatinib bind certain mutated forms of EGFR that frequently occur in lung cancer with higher affinity and are therefore recommended for treatment of these mutated types of non-small cell lung cancer.33,34 Irrespective of the treatment modality, use of EGFR inhibitors (EGFR-I) is associated with 2 common problems, adverse side effects and acquired resistance.5
Side Effects of EGFR-I Treatment
The most common side effects of EGFR-targeted therapies are dermatologic toxicities and diarrhea. Other events, such as nausea, emesis, neurological, or hematologic side effects, are rare compared to cytotoxic chemotherapy.34 Among the cutaneous toxicities observed in cancer patients treated with EGFR-I the most common are papulopustular rash of the upper trunk and face skin (60–90%), dry and itchy skin (12–16%), and microbial infections (38–70%).35,36 Less frequently, pruritus, hair modifications, and paronychial inflammation can occur. The cutaneous side effects seem to be largely independent of the type of EGFR-I used and combined treatment with the monoclonal antibody cetuximab and the TKI erlotinib lead to side effects similar to those induced by treatment with the individual drugs.37 Although side effects induced by EGFR-I are generally classified as moderate, they are usually chronic and may significantly impact the patient's quality of life and thus necessitate dose reduction or even interruption of treatment.
Most importantly, the severity and the timing of the onset of the skin rash significantly correlate with the effectiveness of the treatment.38,39 Correlation was reported when either TKIs or monoclonal antibodies were used as monotherapy or in combination with chemotherapy.39-41 Importantly, rapid onset of moderate to severe skin rash is the best biomarker currently available to predict the effectiveness of EGFR-I treatment, resulting in improved tumor progression parameters such as time to tumor progression and overall survival.42 Meta-analysis revealed that there are minor differences in the response efficiency between ethnic groups and between the individual drugs.40 At present, it is believed that the cutaneous side effects induced by EGFR-I are due to inhibition of EGFR in basal keratinocytes and hair follicles, which express high levels of EGFR similar to tumor cells. Indeed, we and others have recently discovered that lack of EGFR in murine keratinocytes is sufficient to induce skin alterations similar to those observed in the skin of EGFR-I–treated patients, supporting this hypothesis.16,17 However, given that EGFR plays a central role in skin biology, it is currently unclear why the skin rash does not develop in every patient treated with EGFR-I. It has been shown that the severity of the rash is dependent on the dose of EGFR-I used43. Increasing the dose of EGFR-I in patients who do not develop the skin rash at the standard dose was shown to increase skin toxicity along with therapy response rates.38
Recent findings in animal models have shed light on a previously underestimated role of EGFR in immune cells that might be targeted by systemic EGFR-I.44-46 Based on these findings, in the following sections we will discuss the large body of evidence for a central role of EGFR signaling in keratinocytes to maintain homeostasis in the skin and other potential mechanisms underlying the side effects of EGFR-I treatment.
Mechanisms Underlying EGFR-I Induced Cutaneous Side Effects
Skin inflammation, or rash and folliculitis
The skin contains a network of immune cell populations summarized as skin-associated lymphoid tissue (SALT) residing in both the epidermis and the dermis. The cells of this system face the challenge of fulfilling 2 apparently contradictory tasks: on the one hand, to maintain homeostasis and tolerance although confronted with the vast number of microbes that are part of the normal flora, and on the other hand to be prepared to fight against pathogens. The epidermis contains specialized dendritic cells, the Langerhans cells, as well as αβ T-cells and γδ T-cells (also called dendritic epidermal T cells or DETCs), which are rare in human skin but the main T-cell population in mouse epidermis.47 The dermis contains various subpopulations of dendritic cells, macrophages, mast cells, and innate lymphoid cells as well as αβ T cells and γδ T cells. Some of these cell types, like Langerhans cells, constantly migrate to skin-draining lymph nodes under homeostatic conditions whereas other cell types, like neutrophil granulocytes or monocytes, are recruited to the skin to contribute to specific immune responses.48
Skin rash is the most frequently observed side effect in EGFR-I–treated patients, macroscopically appearing with inflammatory papulopustular eruptions that usually start in the face and upper trunk. The rash typically develops within 1 or 2 weeks after therapy initiation, peaks after 2–4 weeks, and slowly regresses with continuation of therapy.49 Histological analysis of skin rash showed mainly CD4-positive T cells and CD1a-positive Langerhans cells throughout the dermis and epidermis, whereas the lesional dermis was dominated by mononuclear myeloid cells like macrophages and activated dendritic cells. Of note, neutrophils were predominantly located at distorted hair follicles.50-54 In mice deficient for Egfr in basal epidermal keratinocytes (EgfrdEP) the resident immune cell populations of the epidermis, Langerhans cells and γδ T-cells, progressively disappeared from the epidermis and were replaced by inflammatory DC and αβ T-cell populations, whereas in the dermis mainly macrophages and mast cells accumulated.16,17 Increased expression of the death receptor ligand TRAIL in cells infiltrating into the dermis that might contribute to rash development has been reported.52 The inflammatory infiltrate is likely triggered by primary changes in epidermal epithelial cells. It may, however, be amplified and maintained by secondary infections and barrier defects, as discussed later in the text. To date, the exact cell type triggering inflammation remains elusive. Application of EGFR-I inhibits EGFR activation in basal cells of the interfollicular epidermis, hair follicle, and eccrine glands.55 Indication for a central role of hair follicle cells in the pathogenesis of skin rash comes from the observation that patients treated with EGFR-I after radiation therapy failed to develop skin rash in irradiated skin areas.56-58 Indeed, hair follicle cells have been reported to respond to radiation by apoptosis whereas epidermal cells responded with cell cycle arrest and temporal depletion of basal layer stem cells.59,60 An alternative hypothesis suggests that depletion of immune cells such as DCs from the skin may be the cause of the transient absence of lesions in previously irradiated skin.61 On the other hand, simultaneous administration of cetuximab in combination with radiation therapy may lead to very severe skin toxicity because of the important role of EGFR in the repair of radiation-induced DNA damage.62-64
Increased expression of CCL-2, CCL-5, and CXCL-10 has been described in EGFR-I–treated cultured human keratinocytes and lesional epidermis.65 Moreover, in interfollicular epidermis, massive production of CCL-27 and CXCL-14 was found that might be responsible for recruitment of T cells and monocyte-derived inflammatory cells, respectively, into the skin (Fig. 2A).16,65 However, high levels of CCL27 detectable in patient serum did not correlate with rash severity.
Figure 2.
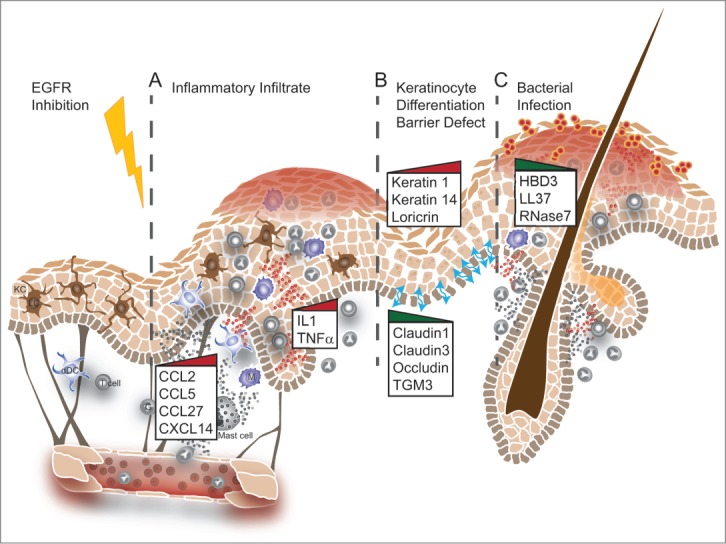
Schematic representation of potential defects observed in the skin of EGFR-I–treated patients. (A) Alterations in chemokine and cytokine production in keratinocytes may result in attraction of inflammatory cells. (B) Disturbed keratinocyte differentiation impairs proper formation of tight junctions and barrier function. (C) The barrier defect and reduced expression of antimicrobial peptides result in bacterial infections accompanied by massive infiltration of neutrophil granulocytes and macrophages. These events often appear sequentially but may also occur independently. The graphic representation is modified from Lacouture M., Rodeck U., (DOI: 10.1126/scitranslmed.3006993)
Levels of other chemokines that are under the control of EGFR ligands in inflamed skin, for example CXCL-1, CXCL-2, CCL-20, CXCL-8, and GM-CSF, are reduced in EGFR-I–treated epidermis.16,65,66 GM-CSF has pleiotropic effects and is not only responsible for differentiation and recruitment of myeloid cells but is also capable of regulating keratinocyte proliferation and apoptosis.67 Similar to EGFR ligands, GM-CSF produced by keratinocytes has been shown to enhance wound healing.68 Little is currently known about the regulatory mechanisms that lead to these changes in cytokine and chemokine expression. GM-CSF seems to be expressed in an ERK/AP1-dependent manner in vitro and in vivo and this regulation is amplified under inflammatory conditions.66 However, in mice some chemokines such as CCL-2 are already upregulated during late embryogenesis, under sterile conditions, and in the absence of detectable inflammatory infiltrate, pointing to a role as transcriptional targets of EGFR. Recent findings indicate another mechanism that might be responsible for the recruitment of neutrophils in particular: treatment of human, but not mouse, keratinocytes with EGFR-I induced expression of complement components and complement activation, resulting in increased deposition of the complement component C3 that might also act as a chemotactic factor.69
Although T cells are abundantly present in the immune infiltrate in lesional skin of patients treated with EGFR-I, experiments in mice lacking Egfr in the epidermis showed that lack of B cells and T cells did not ameliorate the skin inflammation and also did not reduce the immune cell infiltrate, suggesting that the rash might be driven by an innate immune response.16,17 Therefore, therapeutic strategies targeting innate immune cells might be more effective for the management of skin rash in EGFR-I–treated patients. In human skin, blocking IL-8 reduced the side effects induced by locally injected anti-EGFR antibody by reducing infiltration of neutrophils.70 In EgfrdEP mice, local deletion of macrophages or inhibition of mast cell activation reduced skin inflammation and altered the composition of the skin infiltrate, respectively.16,17 Interleukin-1 (IL-1) and its superfamily members IL-18 and IL-33 are candidate cytokines involved in the activation of innate immune responses and rash development. Indeed, IL-1R/MyD88-dependent signaling has been implicated in the induction of sterile inflammation (inflammation occurring in the absence of pathogens) in response to cell death.71 IL-1α is also important for the initiation of the wound healing response that might be responsible for generating an immune cell infiltrate.72,73 Increased expression of IL1α and IL-1β is indeed found in epidermal lysates of EgfrdEP mice, but is probably derived from immune cells since keratinocytes lacking EGFR expressed lower mRNA levels of both cytokines.16 Interestingly, patients with low serum levels of IL-18 before the start of treatment were more likely to develop a higher grade (grade 2 or above) rash than those with high serum levels;17 however, during treatment IL-18 serum levels increased in both patient groups. IL-33, another IL-1 family cytokine, is induced in mouse or human skin after irradiation with UV-B, contributing to the development of inflammation.74 Application of sunscreen can reduce the side effects of EGFR-I in patients, suggesting that sun exposure might potentiate cytokine production in conjunction with reduced EGFR signaling.75
The increased levels of IL1 in EgfrdEP mouse skin, together with the increased IL18 in EGFR-I treated patient serum, suggest that strategies targeting this pathway might be of therapeutic benefit. In a mouse model of neutrophil-rich hair follicle inflammation induced by injection of an anti-mouse Egfr antibody, skin inflammation could be prevented when the IL-1 antagonist kineret was co-injected with the anti-EGFR antibody. However, kineret injection had no effect when injected during ongoing inflammation.76 IL-1 expression seems to be regulated downstream of TNFα since in the same study treatment with etanercept had comparable effects to kineret and reduced IL-1 expression. Discouraging results concerning the inhibition of these proinflammatory pathways come from the analysis of EgfrdEP mice lacking either IL-1 or TNF signaling components. Genetic deletion of MyD88, an adaptor protein necessary for signaling via IL1R and IL18R, did not rescue the inflammatory skin phenotype in mice lacking EGFR in the epidermis and neither did combined deletion of both TNF receptors TNFR1 and TNFR 2.16,17
Barrier defect/xerosis
A major function of the skin is to protect the organism from the environment and from excessive water loss. To fulfill this task, proper differentiation of keratinocytes to form the stratified epithelium and cornification of the outmost layers is necessary. The human interfollicular epidermis consists of 4 layers, the basal layer, spinous layer, granular layer and cornified layer, each of which is characterized by a specific expression pattern of structural and matrix proteins like keratins.77 During terminal differentiation cells become enucleated, involucrin is degraded, the content of lamellar bodies is extruded, and transglutaminases crosslink keratins and other proteins.78 Strong intercellular interactions are formed by tight junction proteins and the lipids in the intercorneocyte space.
Patients receiving EGFR-I can develop xerosis within weeks after treatment start, manifesting as cutaneous dryness, itching, and scaling that are typically found on the limbs and in areas affected by rash but may affect all areas of the skin.53 Mice lacking EGFR signaling in the epidermis develop progressive transepidermal water loss starting around day 10 after birth.16,79 The barrier function of the skin may be perturbed by EGFR-I treatment at multiple levels (Fig. 2B). EGFR ligands like EGF or TGFα induce proliferation of cultured keratinocytes and epidermal hyperplasia and hyperkeratosis,80,81 whereas in benign papilloma EGFR has been shown to provide a survival signal to keratinocytes.82
Furthermore, EGFR signaling regulates differentiation of keratinocytes and the activity of transglutaminases (TGM) that are critical for crosslinking structural proteins like involucrin, loricrin, and small proline-rich proteins by forming 3-(γ-glutamyl) lysine isopeptide bonds. In EgfrdEP mice and in Adam17ΔKC mice the expression of tgm3 is strongly reduced accompanied by altered epidermal differentiation, mirrored by retained expression of loricrin in basal keratinocytes and a derangement of keratin expression.16,79 TGFα produced by keratinocytes is the main EGFR ligand responsible for barrier integrity since mice lacking tgfα develop a similar phenotype.83,84 This might in part be mediated by Ca2+ signaling. The calcium channel transient receptor potential 3 (trpv3) protein has recently been shown to be an essential part of the EGFR signaling cascade. Lack of trpv3 in the epidermis affects hair development and skin barrier function and results in a phenotype similar to that of waved (wa)1 and wa2 mice, which are either deficient for TGFα (wa1) or harbor a hypomorphic EGFR variant (wa2).83,84,109 Calcium influx via trpv3 induces further transactivation of EGFR and is necessary for transglutaminase activity in the superficial epidermis. Consequently, changes in stratification, parakeratosis (the retention of nuclei in stratum corneum), vacuolar degeneration, and apoptotic keratinocytes in the basal layer can be observed in EGFR-I–treated patients.55
EGFR is also implicated in regulating cell–cell contacts, including those in cancer cells. Treatment of the A431 tumor cell line with EGFR-I decreased expression of claudin-4, and increased claudin-2 expression in the human lung adenocarcinoma cell line A549 was dependent on EGFR activation.85,86 Expression of claudin-3 was reduced in EgfrdEP epidermis, as was expression of claudin-1 in human lesional skin (Fig. 2B).16 Therefore, skin barrier defects can lead to compensatory hyperproliferation, as indicated by increased proliferation and keratin 6 expression, and might be responsible for some of the proinflammatory factors secreted by lesional skin as well as the production of “danger” cytokines like IL-36.79,87
Antimicrobial defense/infections
The skin not only constitutes a physical barrier protecting from infection but is also an environmental niche hosting a plethora of commensal organisms such as bacteria and fungi. These microorganisms are specifically adapted for this niche and protect the body by preventing colonization and invasion of opportunistic organisms.88 Defects in epidermal structural proteins may disturb this barrier function. For example, mutations of filaggrin are associated with atopic dermatitis, and a shift in skin microbiota has been observed in a related mouse model with abnormal filaggrin processing.89 Combined with the physical epidermal barrier, antimicrobial peptides (AMPs) constitute a major component of the active innate immune defense against invading microbes in the skin. Under homeostatic conditions AMPs in the skin are produced mainly by keratinocytes, but also by mast cells and eccrine sweat glands.90 Only recently it has become understood that a substantial contribution also comes from commensal bacteria, which produce AMPs and also TLR ligands.91,92 Under inflammatory conditions, a large number of “inducible” AMPs are produced by infiltrating immune cells like neutrophils or dendritic cells. The basic functions of all AMPs are activation of the host innate immune response and direct killing of pathogens.93 The major groups of AMPs found in the skin are defensins, cathelicidin, dermcidin, and a group of other proteins/peptides including RNAse7 and S100 proteins.
Defensins have a broad spectrum of antimicrobial activity and their expression can be induced by bacterial infection or proinflammatory cytokines like IL-1, but may be inhibited by pretreatment with retinoic acid.93 Cathelicidin (CAMP, LL-37) has broad-spectrum activity and has been shown to bind to lipopolysaccharide (LPS) on gram-negative bacteria but also activates keratinocytes. Skin inflammation and 1,25-dehydroxy vitamin D3 are potent inducers of cathelicidin expression.93 Expression of some AMPs is also under the control of EGFR. Bacterial products like the Helicobacter pylori virulence effector CagA or LPS function via EGFR signaling to either suppress or upregulate expression of human β-defensin 3, respectively.94,95 Treatment of human keratinocytes with erlotinib reduced expression of hBD3, RNase7, and CAMP; similarly, expression of murine β-defensin 14, the mouse homolog of hBD3, was reduced in egfr-deficient keratinocytes (Fig. 2C).16 However, expression of β-defensin 1 and 2 was increased in these cells, which is consistent with reports of differential regulation of expression of hBD1-4 in humans.93 EGFR also closely interacts with S100 proteins and regulates the transcription of S100A2 and S100A7 (Psoriasin).96,97 S100A7 protein has been shown to interfere with EGFR signaling and increase survival of cancer cell lines, whereas S100A4 protein binds to EGFR ligands to enhance proliferation.98,99 In normal human keratinocytes, induction of CCL20, hBD4, and S100A7 RNA expression requires the synergistic action of integrins, EGFR, and IL-1 to promote antimicrobial defense.100
The question of whether treatment with EGFR-I affects antimicrobial defense has already been addressed in early clinical trials. The folliculitis observed in EGFR-I–treated patients typically begins as a sterile inflammatory process associated with neutrophil-rich infiltrates, which is in contrast to classic acne.25,54 Acne is believed to be caused by infection with Propionibacterium acnes and colonization of lesions with this bacterium can be found at day 1 in 70% of patients.101 Interestingly, EGFR-I–treated patients showed no significant changes in the cutaneous microflora at early stages, although Staphylococcus aureus was cultured from some persistent lesions.51,52 More recent studies revealed that up to 40% of patients developed infections after EGFR-I treatment, mainly at sites of toxic lesions, with the vast majority of infections being caused by S. aureus and fewer by infection with enterobacteriaceae and herpes viridae.16,102 That the increased rate of infections might indeed be the result of reduced AMP production by keratinocytes is indicated by the finding that supernatants from erlotinib-treated human keratinocytes were less potent in killing S. aureus than those from mock-treated control keratinocytes.16 Infection with pathogenic strains like S. aureus may worsen the course of EGFR-I–induced cutaneous disease and thus further compromise the patient's quality of life, creating the need for effective management of complicating infections. Antibiotics of the tetracycline family, namely doxycycline and minocycline, have recently been evaluated for their potential to alleviate EGFR-I–induced skin disease. In one study, combined treatment with moisture, topical sun screen, prednisolone, and doxycycline from day 1 before the start of EGFR-I and continuing over a 6-week period resulted in a significant reduction in the number of patients with rash of grade >2.75 Another study that evaluated the use of doxycycline alone observed a lower incidence of grade 2–3 folliculitis in the tetracycline arm.49 Thus, preventive application of tetracyclines seems to be a promising approach to improve the quality of life that is affected by the impaired antimicrobial defense caused by EGFR-I treatment. Tetracyclines are also applied in a variety of dermatologic diseases because of their anti-inflammatory activity through inhibiting cytokine and chemokine secretion.49,103 Doxycycline has already been shown to be effective in rosacea therapy at sub-antimicrobial doses.104 Furthermore, high rates of tetracycline-resistant S. aureus are found in lesions of EGFR-I patients, suggesting that the ameliorative effects of preemptive tetracycline therapy on EGFR-I induced skin rash are due to its anti-inflammatory properties.102
Hair alterations
Hair modifications in EGFR-I–treated patients develop at later time points than folliculitis, usually after 2–3 months, and differentially affect distinct hair types. Although scalp hair grows more slowly and some patients eventually develop androgen-like alopecia, facial hair and eyelashes may grow progressively.35,36
Hair follicles are constantly self-renewing throughout life, a process called the hair cycle. The hair cycle consists of 3 stages: anagen (hair growth), catagen (phase of involution), and telogen (the resting phase). The EGFR ligands EGF and TGFα play a critical role during the hair cycle because they are involved in triggering anagen and catagen phases and in the differentiation of sebocytes.105,106 The threshold of EGFR activation during hair growth and development seems to be very important, as hyper- and hypoactivation of the EGFR can lead to a similar outcome. Overexpression of EGF in mice results in thinner hair, and EGF treatment of newborn mice delays hair development.107,108 Moreover, mice harboring a hypomorphic EGFR (wa2) develop wavy hair and curly whiskers, as do mice harboring a mutated form (wa1) or homozygous deletion of TGFα.83,84,109 Studies in different EGFR mutant mouse models showed that, after the first hair cycle, EGFR-deficient hair follicles fail to enter catagen and remain in aberrant anagen, which results in hair follicle degradation and hair loss.18,110 These changes are accompanied by fibrosis and an inflammatory infiltrate. The complex functions of EGFR in the epidermis and hair follicles are also highlighted by transplantation experiments with EGFR-deficient skin. A block in hair cycle progression was observed when EGFR-deficient skin was grafted onto wild-type mice, suggesting a cell-autonomous requirement for EGFR.111 However, progressive hair follicle loss in these transplants was always accompanied by a dense dermal immune infiltrate. Whether similar mechanisms cause hair alterations in EGFR-I–treated patients remains to be determined. Since immune cell infiltration and hair loss are also always associated in EGFR-I–treated patients it is likely that hair loss is a result of immune-mediated damage, suggesting that EGFR signaling might be protective against immunological reactions.18,111
Some studies also point toward an involvement of hormone signaling in EGFR-I–induced alopecia. Indeed, data from breast cancer studies indicate interactions between hormone receptors and EGFR.112,113 Additionally, crosstalk between EGFR and the estrogen receptor pathway has been reported in squamous cell carcinoma.114 Some data indicate that trichomegaly preferentially occurs in female patients.115 However, careful analyses of the hormone pathways in treated patients are still lacking.
Conclusion
Our understanding of the pathogenesis underlying EGFR-I–induced skin toxicities has substantially increased during the last years due in part to the generation of novel animal models addressing the specific role of EGFR and its ligands in keratinocytes. Clinical studies have evaluated new treatment regimens that led to novel guidelines for the treatment of patients receiving EGFR-I, involving pre-emptive treatment with antibiotics and intensive skin care.75 However, more prospective clinical studies are needed to optimize these treatment strategies. Additionally, evidence-based approaches must be established. One example would be treatment with inhibitors of IL-1, based on its high expression in the epidermis. However, with the increasing number of clinical and preclinical studies it has become evident that systemic treatment strategies might also reduce the effectiveness of EGFR-I against tumor growth that seems to be mediated in part by an antitumor immune response.116-119 Therefore, novel topical strategies to reduce side effects in the skin are needed. Case reports of topical recombinant human EGF or topical vitamin K cream resulting in a reduction of rash grade within a few weeks are very promising.120,121 Menadione, a synthetic pro-vitamin K3, inhibits phosphatases and thereby increases baseline EGFR phosphorylation.122 However, topical pretreatment with vitamin K1 (phytomenadione) did not profoundly affect rash severity.123 With EGFR-I treatment being applied more and more in early stages of antitumor therapy, the need for strategies to reduce its side effects will further increase to avoid dose reductions and maintain patients on therapy at an effective dose. Future preselection strategies for patients should consider testing skin biopsies for the positive development of skin rash in vitro in organ cultures to predict the patient's response to anti-EGFR therapies. This would certainly increase the treatment success rate and avoid unnecessary treatments and related costs. Only then we will be able to exploit the full potential of anti-EGFR therapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Gerald Prager for critical reading of the manuscript and Dr. Thomas Bauer for the artwork (design of Figs. 1 and 2).
References
- 1.Atlas I, Mendelsohn J, Baselga J, Fair WR, Masui H, Kumar R. Growth regulation of human renal carcinoma cells: role of transforming growth factor alpha. Cancer Res 1992; 52:3335–9; PMID:1596891 [PubMed] [Google Scholar]
- 2.Wu X, Rubin M, Fan Z, DeBlasio T, Soos T, Koff A, Mendelsohn J. Involvement of p27KIP1 in G1 arrest mediated by an anti-epidermal growth factor receptor monoclonal antibody. Oncogene 1996; 12:1397–403; PMID:8622855 [PubMed] [Google Scholar]
- 3.Bruns CJ, Harbison MT, Davis DW, Portera CA, Tsan R, McConkey DJ, Evans DB, Abbruzzese JL, Hicklin DJ, Radinsky R. Epidermal growth factor receptor blockade with C225 plus gemcitabine results in regression of human pancreatic carcinoma growing orthotopically in nude mice by antiangiogenic mechanisms. Clin Cancer Res 2000; 6:1936–48; PMID:10815919 [PubMed] [Google Scholar]
- 4.Petit AM, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B, Kerbel RS. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol 1997; 151:1523–30; PMID:9403702 [PMC free article] [PubMed] [Google Scholar]
- 5.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013; 19:1389–400; PMID:24202392; http://dx.doi.org/ 10.1038/nm.3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J Cell Physiol 2009; 218:460–6; PMID:19006176; http://dx.doi.org/ 10.1002/jcp.21635 [DOI] [PubMed] [Google Scholar]
- 7.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 2004; 164:769–79; PMID:14993236; http://dx.doi.org/ 10.1083/jcb.200307137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh AB, Harris RC. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal 2005; 17:1183–93; PMID:15982853; http://dx.doi.org/ 10.1016/j.cellsig.2005.03.026 [DOI] [PubMed] [Google Scholar]
- 9.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2:127–37; PMID:11252954; http://dx.doi.org/ 10.1038/35052073 [DOI] [PubMed] [Google Scholar]
- 10.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995; 376:337–41; PMID:7630400; http://dx.doi.org/ 10.1038/376337a0 [DOI] [PubMed] [Google Scholar]
- 11.Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A 2007; 104:17081–6; PMID:17940036; http://dx.doi.org/ 10.1073/pnas.0704126104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 2007; 75:770–87; PMID:17999740; http://dx.doi.org/ 10.1111/j.1432-0436.2007.00238.x [DOI] [PubMed] [Google Scholar]
- 13.Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995; 269:234–8; PMID:7618085; http://dx.doi.org/ 10.1126/science.7618085 [DOI] [PubMed] [Google Scholar]
- 14.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al.. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 1995; 269:230–4; PMID:7618084; http://dx.doi.org/ 10.1126/science.7618084 [DOI] [PubMed] [Google Scholar]
- 15.Sibilia M, Steinbach JP, Stingl L, Aguzzi A, Wagner EF. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J 1998; 17:719–31; PMID:9450997; http://dx.doi.org/ 10.1093/emboj/17.3.719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lichtenberger BM, Gerber PA, Holcmann M, Buhren BA, Amberg N, Smolle V, Schrumpf H, Boelke E, Ansari P, Mackenzie C, et al.. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci Transl Med 2013; 5:199ra11; http://dx.doi.org/ 10.1126/scitranslmed.3005886 [DOI] [PubMed] [Google Scholar]
- 17.Mascia F, Lam G, Keith C, Garber C, Steinberg SM, Kohn E, Yuspa SH. Genetic ablation of epidermal EGFR reveals the dynamic origin of adverse effects of anti-EGFR therapy. Sci Transl Med 2013; 5:199ra10; http://dx.doi.org/ 10.1126/scitranslmed.3005773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sibilia M, Wagner B, Hoebertz A, Elliott C, Marino S, Jochum W, Wagner EF. Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development 2003; 130:4515–25; PMID:12925580; http://dx.doi.org/ 10.1242/dev.00664 [DOI] [PubMed] [Google Scholar]
- 19.Campbell P, Morton PE, Takeichi T, Salam A, Roberts N, Proudfoot LE, Mellerio JE, Aminu K, Wellington C, Patil SN, et al.. Epithelial Inflammation resulting from an inherited loss-of-function mutation in EGFR. J Invest Dermatol 2014; 134:2570–8; PMID:24691054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Na II, Kang HJ, Cho SY, Koh JS, Lee JK, Lee BC, Lee GH, Lee YS, Yoo HJ, Ryoo BY, et al.. EGFR mutations and human papillomavirus in squamous cell carcinoma of tongue and tonsil. Eur J Cancer 2007; 43:520–6; PMID:17224267; http://dx.doi.org/ 10.1016/j.ejca.2006.09.025 [DOI] [PubMed] [Google Scholar]
- 21.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001; 37 Suppl 4:S9–15; PMID:11597399; http://dx.doi.org/ 10.1016/S0959-8049(01)00231-3 [DOI] [PubMed] [Google Scholar]
- 22.Sihto H, Puputti M, Pulli L, Tynninen O, Koskinen W, Aaltonen LM, Tanner M, Bohling T, Visakorpi T, Butzow R, et al.. Epidermal growth factor receptor domain II, IV, and kinase domain mutations in human solid tumors. J Mol Med (Berl) 2005; 83:976–83; PMID:16133419; http://dx.doi.org/ 10.1007/s00109-005-0699-4 [DOI] [PubMed] [Google Scholar]
- 23.Huang SM, Harari PM. Epidermal growth factor receptor inhibition in cancer therapy: biology, rationale and preliminary clinical results. Invest New Drugs 1999; 17:259–69; PMID:10665478; http://dx.doi.org/ 10.1023/A:1006384521198 [DOI] [PubMed] [Google Scholar]
- 24.Masui H, Kawamoto T, Sato JD, Wolf B, Sato G, Mendelsohn J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res 1984; 44:1002–7; PMID:6318979 [PubMed] [Google Scholar]
- 25.Baselga J. The EGFR as a target for anticancer therapy–focus on cetuximab. Eur J Cancer 2001; 37 Suppl 4:S16–22; PMID:11597400; http://dx.doi.org/ 10.1016/S0959-8049(01)00233-7 [DOI] [PubMed] [Google Scholar]
- 26.Haraldsdottir S, Bekaii-Saab T. Integrating anti-EGFR therapies in metastatic colorectal cancer. J Gastrointest Oncol 2013; 4:285–98; PMID:23997940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol 2012; 30:3570–7; PMID:22734028; http://dx.doi.org/ 10.1200/JCO.2012.42.2592 [DOI] [PubMed] [Google Scholar]
- 28.Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, et al.. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 2009; 27:5068–74; PMID:19738126; http://dx.doi.org/ 10.1200/JCO.2008.21.3744 [DOI] [PubMed] [Google Scholar]
- 29.Pentheroudakis G, Kotoula V, De Roock W, Kouvatseas G, Papakostas P, Makatsoris T, Papamichael D, Xanthakis I, Sgouros J, Televantou D, et al.. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC Cancer 2013; 13:49; PMID:23374602; http://dx.doi.org/ 10.1186/1471-2407-13-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Einem JC, Heinemann V, von Weikersthal LF, Vehling-Kaiser U, Stauch M, Hass HG, Decker T, Klein S, Held S, Jung A, et al.. Left-sided primary tumors are associated with favorable prognosis in patients with KRAS codon 12/13 wild-type metastatic colorectal cancer treated with cetuximab plus chemotherapy: an analysis of the AIO KRK-0104 trial. J Cancer Res Clin Oncol 2014; 140:1607–14; PMID:24816724; http://dx.doi.org/ 10.1007/s00432-014-1678-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith CG, Fisher D, Claes B, Maughan TS, Idziaszczyk S, Peuteman G, Harris R, James MD, Meade A, Jasani B, et al.. Somatic profiling of the epidermal growth factor receptor pathway in tumors from patients with advanced colorectal cancer treated with chemotherapy +/− cetuximab. Clin Cancer Res 2013; 19:4104–13; PMID:23741067; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sebio A, Paez D, Salazar J, Berenguer-Llergo A, Pare-Brunet L, Lasa A, Del Rio E, Tobena M, Martin-Richard M, Baiget M, et al.. Intergenic polymorphisms in the amphiregulin gene region as biomarkers in metastatic colorectal cancer patients treated with anti-EGFR plus irinotecan. Pharmacogenomics J 2014; 14:256–62; PMID:23959273; http://dx.doi.org/ 10.1038/tpj.2013.29 [DOI] [PubMed] [Google Scholar]
- 33.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008; 358:1160–74; PMID:18337605; http://dx.doi.org/ 10.1056/NEJMra0707704 [DOI] [PubMed] [Google Scholar]
- 34.Sanford M, Scott LJ. Gefitinib: a review of its use in the treatment of locally advanced/metastatic non-small cell lung cancer. Drugs 2009; 69:2303–28; PMID:19852530; http://dx.doi.org/ 10.2165/10489100-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 35.Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer 2006; 6:803–12; PMID:16990857; http://dx.doi.org/ 10.1038/nrc1970 [DOI] [PubMed] [Google Scholar]
- 36.Robert C, Soria JC, Spatz A, Le Cesne A, Malka D, Pautier P, Wechsler J, Lhomme C, Escudier B, Boige V, et al.. Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol 2005; 6:491–500; PMID:15992698; http://dx.doi.org/ 10.1016/S1470-2045(05)70243-6 [DOI] [PubMed] [Google Scholar]
- 37.Guarino MJ, Schneider CJ, Hosford MA, Brahmer JR, Rudin CM, Finckenstein FG, Philip-Norton RE, Lu H, Weber MR, Ettinger DS. Dual inhibition of the epidermal growth factor receptor pathway with cetuximab and erlotinib: a phase I study in patients with advanced solid malignancies. Oncologist 2009; 14:119–24; PMID:19182243; http://dx.doi.org/ 10.1634/theoncologist.2008-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Cutsem E, Tejpar S, Vanbeckevoort D, Peeters M, Humblet Y, Gelderblom H, Vermorken JB, Viret F, Glimelius B, Gallerani E, et al.. Intrapatient cetuximab dose escalation in metastatic colorectal cancer according to the grade of early skin reactions: the randomized EVEREST study. J Clin Oncol 2012; 30:2861–8; PMID:22753904; http://dx.doi.org/ 10.1200/JCO.2011.40.9243 [DOI] [PubMed] [Google Scholar]
- 39.Wacker B, Nagrani T, Weinberg J, Witt K, Clark G, Cagnoni PJ. Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin Cancer Res 2007; 13:3913–21; PMID:17606725; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-2610 [DOI] [PubMed] [Google Scholar]
- 40.Liu HB, Wu Y, Lv TF, Yao YW, Xiao YY, Yuan DM, Song Y. Skin rash could predict the response to EGFR tyrosine kinase inhibitor and the prognosis for patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS One 2013; 8:e55128; PMID:23383079; http://dx.doi.org/ 10.1371/journal.pone.0055128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Soler R, Saltz L. Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol 2005; 23:5235–46; PMID:16051966; http://dx.doi.org/ 10.1200/JCO.2005.00.6916 [DOI] [PubMed] [Google Scholar]
- 42.Saridaki Z, Tzardi M, Papadaki C, Sfakianaki M, Pega F, Kalikaki A, Tsakalaki E, Trypaki M, Messaritakis I, Stathopoulos E, et al.. Impact of KRAS, BRAF, PIK3CA mutations, PTEN, AREG, EREG expression and skin rash in >/= 2 line cetuximab-based therapy of colorectal cancer patients. PLoS One 2011; 6:e15980; PMID:21283802; http://dx.doi.org/ 10.1371/journal.pone.0015980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fakih M, Vincent M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr Oncol 2010; 17 Suppl 1:S18–30; PMID:20680104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lanaya H, Natarajan A, Komposch K, Li L, Amberg N, Chen L, Wculek SK, Hammer M, Zenz R, Peck-Radosavljevic M, et al.. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol 2014; 16:972–81; PMID:25173978; http://dx.doi.org/ 10.1038/ncb3031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu N, Wang L, Cao H, Liu L, Van Kaer L, Washington MK, Rosen MJ, Dube PE, Wilson KT, Ren X, et al.. Activation of the epidermal growth factor receptor in macrophages regulates cytokine production and experimental colitis. J Immunol 2014; 192:1013–23; http://dx.doi.org/ 10.4049/jimmunol.1300133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Grone A, Sibilia M, van Bergen en Henegouwen PM, Roovers RC, Coffer PJ, Sijts AJ. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity 2013; 38:275–84; PMID:23333074; http://dx.doi.org/ 10.1016/j.immuni.2012.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tay SS, Roediger B, Tong PL, Tikoo S, Weninger W. The skin-resident immune network. Curr Dermatol Rep 2014; 3:13–22; PMID:24587975; http://dx.doi.org/ 10.1007/s13671-013-0063-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heath WR, Carbone FR. The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol 2013; 14:978–85; PMID:24048119; http://dx.doi.org/ 10.1038/ni.2680 [DOI] [PubMed] [Google Scholar]
- 49.Scope A, Agero AL, Dusza SW, Myskowski PL, Lieb JA, Saltz L, Kemeny NE, Halpern AC. Randomized double-blind trial of prophylactic oral minocycline and topical tazarotene for cetuximab-associated acne-like eruption. J Clin Oncol 2007; 25:5390–6; PMID:18048820; http://dx.doi.org/ 10.1200/JCO.2007.12.6987 [DOI] [PubMed] [Google Scholar]
- 50.Brodell LA, Hepper D, Lind A, Gru AA, Anadkat MJ. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol 2013; 40:865–70; PMID:23941617 [DOI] [PubMed] [Google Scholar]
- 51.Busam KJ, Capodieci P, Motzer R, Kiehn T, Phelan D, Halpern AC. Cutaneous side-effects in cancer patients treated with the antiepidermal growth factor receptor antibody C225. Br J Dermatol 2001; 144:1169–76; PMID:11422037; http://dx.doi.org/ 10.1046/j.1365-2133.2001.04226.x [DOI] [PubMed] [Google Scholar]
- 52.Guttman-Yassky E, Mita A, De Jonge M, Matthews L, McCarthy S, Iwata KK, Verweij J, Rowinsky EK, Krueger JG. Characterisation of the cutaneous pathology in non-small cell lung cancer (NSCLC) patients treated with the EGFR tyrosine kinase inhibitor erlotinib. Eur J Cancer 2010; 46:2010–9; PMID:20621734; http://dx.doi.org/ 10.1016/j.ejca.2010.04.028 [DOI] [PubMed] [Google Scholar]
- 53.Segaert S, Van Cutsem E. Clinical signs, pathophysiology and management of skin toxicity during therapy with epidermal growth factor receptor inhibitors. Ann Oncol 2005; 16:1425–33; PMID:16012181; http://dx.doi.org/ 10.1093/annonc/mdi279 [DOI] [PubMed] [Google Scholar]
- 54.Van Doorn R, Kirtschig G, Scheffer E, Stoof TJ, Giaccone G. Follicular and epidermal alterations in patients treated with ZD1839 (Iressa), an inhibitor of the epidermal growth factor receptor. Br J Dermatol 2002; 147:598–601; PMID:12207609; http://dx.doi.org/ 10.1046/j.1365-2133.2002.04864.x [DOI] [PubMed] [Google Scholar]
- 55.Albanell J, Rojo F, Averbuch S, Feyereislova A, Mascaro JM, Herbst R, LoRusso P, Rischin D, Sauleda S, Gee J, et al.. Pharmacodynamic studies of the epidermal growth factor receptor inhibitor ZD1839 in skin from cancer patients: histopathologic and molecular consequences of receptor inhibition. J Clin Oncol 2002; 20:110–24; PMID:11773160; http://dx.doi.org/ 10.1200/JCO.20.1.110 [DOI] [PubMed] [Google Scholar]
- 56.Acharya J, Lyon C, Bottomley DM. Folliculitis-perifolliculitis related to erlotinib therapy spares previously irradiated skin. J Am Acad Dermatol 2009; 60:154–7; PMID:19103369; http://dx.doi.org/ 10.1016/j.jaad.2008.07.057 [DOI] [PubMed] [Google Scholar]
- 57.Bossi P, Liberatoscioli C, Bergamini C, Locati LD, Fava S, Rinaldi G, Orlandi E, Olmi P, Tagliabue E, Menard S, et al.. Previously irradiated areas spared from skin toxicity induced by cetuximab in six patients: implications for the administration of EGFR inhibitors in previously irradiated patients. Ann Oncol 2007; 18:601–2; PMID:17074970; http://dx.doi.org/ 10.1093/annonc/mdl409 [DOI] [PubMed] [Google Scholar]
- 58.Lacouture ME, Hwang C, Marymont MH, Patel J. Temporal dependence of the effect of radiation on erlotinib-induced skin rash. J Clin Oncol 2007; 25:2140; author reply 1; PMID:17513824; http://dx.doi.org/ 10.1200/JCO.2006.09.4045 [DOI] [PubMed] [Google Scholar]
- 59.Song S, Lambert PF. Different responses of epidermal and hair follicular cells to radiation correlate with distinct patterns of p53 and p21 induction. Am J Pathol 1999; 155:1121–7; PMID:10514395; http://dx.doi.org/ 10.1016/S0002-9440(10)65215-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hymes SR, Strom EA, Fife C. Radiation dermatitis: clinical presentation, pathophysiology, and treatment 2006. J Am Acad Dermatol 2006; 54:28–46; PMID:16384753; http://dx.doi.org/ 10.1016/j.jaad.2005.08.054 [DOI] [PubMed] [Google Scholar]
- 61.Gerber PA, Enderlein E, Homey B, Muller A, Boelke E, Budach W. Radiation-induced prevention of erlotinib-induced skin rash is transient: a new aspect toward the understanding of epidermal growth factor receptor inhibitor associated cutaneous adverse effects. J Clin Oncol 2007; 25:4697–8; author reply 8-9; PMID:17925571; http://dx.doi.org/ 10.1200/JCO.2007.12.8330 [DOI] [PubMed] [Google Scholar]
- 62.Bolke E, Gerber PA, Lammering G, Peiper M, Muller-Homey A, Pape H, Giro C, Matuschek C, Bruch-Gerharz D, Hoffmann TK, et al.. Development and management of severe cutaneous side effects in head-and-neck cancer patients during concurrent radiotherapy and cetuximab. Strahlenther Onkol 2008; 184:105–10; PMID:18259703; http://dx.doi.org/ 10.1007/s00066-008-1829-z [DOI] [PubMed] [Google Scholar]
- 63.Budach W, Bolke E, Homey B. Severe cutaneous reaction during radiation therapy with concurrent cetuximab. N Engl J Med 2007; 357:514–5; PMID:17671265; http://dx.doi.org/ 10.1056/NEJMc071075 [DOI] [PubMed] [Google Scholar]
- 64.Liccardi G, Hartley JA, Hochhauser D. Importance of EGFR/ERCC1 interaction following radiation-induced DNA damage. Clin Cancer Res 2014; 20:3496–506; PMID:24780295; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2695 [DOI] [PubMed] [Google Scholar]
- 65.Mascia F, Mariani V, Girolomoni G, Pastore S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol 2003; 163:303–12; PMID:12819035; http://dx.doi.org/ 10.1016/S0002-9440(10)63654-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mascia F, Cataisson C, Lee TC, Threadgill D, Mariani V, Amerio P, Chandrasekhara C, Souto Adeva G, Girolomoni G, Yuspa SH, et al.. EGFR regulates the expression of keratinocyte-derived granulocyte/macrophage colony-stimulating factor in vitro and in vivo. J Invest Dermatol 2010; 130:682–93; PMID:19890352; http://dx.doi.org/ 10.1038/jid.2009.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Breuhahn K, Mann A, Muller G, Wilhelmi A, Schirmacher P, Enk A, Blessing M. Epidermal overexpression of granulocyte-macrophage colony-stimulating factor induces both keratinocyte proliferation and apoptosis. Cell Growth Differ 2000; 11:111–21; PMID:10714767 [PubMed] [Google Scholar]
- 68.Mann A, Breuhahn K, Schirmacher P, Blessing M. Keratinocyte-derived granulocyte-macrophage colony stimulating factor accelerates wound healing: stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J Invest Dermatol 2001; 117:1382–90; PMID:11886498; http://dx.doi.org/ 10.1046/j.0022-202x.2001.01600.x [DOI] [PubMed] [Google Scholar]
- 69.Abu-Humaidan AH, Ananthoju N, Mohanty T, Sonesson A, Alberius P, Schmidtchen A, Garred P, Sorensen OE. The epidermal growth factor receptor is a regulator of epidermal complement component expression and complement activation. J Immunol 2014; 192:3355–64; http://dx.doi.org/ 10.4049/jimmunol.1302305 [DOI] [PubMed] [Google Scholar]
- 70.Bangsgaard N, Houtkamp M, Schuurhuis DH, Parren PW, Baadsgaard O, Niessen HW, Skov L. Neutralization of IL-8 prevents the induction of dermatologic adverse events associated with the inhibition of epidermal growth factor receptor. PLoS One 2012; 7:e39706; PMID:22761877; http://dx.doi.org/ 10.1371/journal.pone.0039706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13:851–6; PMID:17572686; http://dx.doi.org/ 10.1038/nm1603 [DOI] [PubMed] [Google Scholar]
- 72.Freedberg IM, Tomic-Canic M, Komine M, Blumenberg M. Keratins and the keratinocyte activation cycle. J Invest Dermatol 2001; 116:633–40; PMID:11348449; http://dx.doi.org/ 10.1046/j.1523-1747.2001.01327.x [DOI] [PubMed] [Google Scholar]
- 73.Murphy JE, Robert C, Kupper TS. Interleukin-1 and cutaneous inflammation: a crucial link between innate and acquired immunity. J Invest Dermatol 2000; 114:602–8; PMID:10692124; http://dx.doi.org/ 10.1046/j.1523-1747.2000.00917.x [DOI] [PubMed] [Google Scholar]
- 74.Byrne SN, Beaugie C, O'Sullivan C, Leighton S, Halliday GM. The immune-modulating cytokine and endogenous Alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am J Pathol 2011; 179:211–22; PMID:21703403; http://dx.doi.org/ 10.1016/j.ajpath.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lacouture ME, Mitchell EP, Piperdi B, Pillai MV, Shearer H, Iannotti N, Xu F, Yassine M. Skin toxicity evaluation protocol with panitumumab (STEPP), a phase II, open-label, randomized trial evaluating the impact of a pre-emptive skin treatment regimen on skin toxicities and quality of life in patients with metastatic colorectal cancer. J Clin Oncol 2010; 28:1351–7; PMID:20142600; http://dx.doi.org/ 10.1200/JCO.2008.21.7828 [DOI] [PubMed] [Google Scholar]
- 76.Surguladze D, Deevi D, Claros N, Corcoran E, Wang S, Plym MJ, Wu Y, Doody J, Mauro DJ, Witte L, et al.. Tumor necrosis factor-alpha and interleukin-1 antagonists alleviate inflammatory skin changes associated with epidermal growth factor receptor antibody therapy in mice. Cancer Res 2009; 69:5643–7; PMID:19584274; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0487 [DOI] [PubMed] [Google Scholar]
- 77.Eckhart L, Lippens S, Tschachler E, Declercq W. Cell death by cornification. Biochim Biophys Acta 2013; 1833:3471–80; PMID:23792051; http://dx.doi.org/ 10.1016/j.bbamcr.2013.06.010 [DOI] [PubMed] [Google Scholar]
- 78.Zeeuwen PL. Epidermal differentiation: the role of proteases and their inhibitors. Eur J Cell Biol 2004; 83:761–73; PMID:15679120; http://dx.doi.org/ 10.1078/0171-9335-00388 [DOI] [PubMed] [Google Scholar]
- 79.Franzke CW, Cobzaru C, Triantafyllopoulou A, Loffek S, Horiuchi K, Threadgill DW, Kurz T, van Rooijen N, Bruckner-Tuderman L, Blobel CP. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J Exp Med 2012; 209:1105–19; PMID:22565824; http://dx.doi.org/ 10.1084/jem.20112258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cohen S. The stimulation of epidermal proliferation by a specific protein (EGF). Dev Biol 1965; 12:394–407; PMID:5884352; http://dx.doi.org/ 10.1016/0012-1606(65)90005-9 [DOI] [PubMed] [Google Scholar]
- 81.Dominey AM, Wang XJ, King LE Jr., Nanney LB, Gagne TA, Sellheyer K, Bundman DS, Longley MA, Rothnagel JA, Greenhalgh DA, et al.. Targeted overexpression of transforming growth factor alpha in the epidermis of transgenic mice elicits hyperplasia, hyperkeratosis, and spontaneous, squamous papillomas. Cell Growth Differ 1993; 4:1071–82; PMID:8117621 [PubMed] [Google Scholar]
- 82.Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 2010; 140:268–79; PMID:20141840; http://dx.doi.org/ 10.1016/j.cell.2009.12.046 [DOI] [PubMed] [Google Scholar]
- 83.Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell 1993; 73:263–78; PMID:8477445; http://dx.doi.org/ 10.1016/0092-8674(93)90228-I [DOI] [PubMed] [Google Scholar]
- 84.Mann GB, Fowler KJ, Gabriel A, Nice EC, Williams RL, Dunn AR. Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell 1993; 73:249–61; PMID:8477444; http://dx.doi.org/ 10.1016/0092-8674(93)90227-H [DOI] [PubMed] [Google Scholar]
- 85.Ikari A, Sato T, Watanabe R, Yamazaki Y, Sugatani J. Increase in claudin-2 expression by an EGFR/MEK/ERK/c-Fos pathway in lung adenocarcinoma A549 cells. Biochim Biophys Acta 2012; 1823:1110–8; PMID:22546605; http://dx.doi.org/ 10.1016/j.bbamcr.2012.04.005 [DOI] [PubMed] [Google Scholar]
- 86.Singh AB, Sugimoto K, Dhawan P, Harris RC. Juxtacrine activation of EGFR regulates claudin expression and increases transepithelial resistance. Am J Physiol Cell Physiol 2007; 293:C1660–8; PMID:17855771; http://dx.doi.org/ 10.1152/ajpcell.00274.2007 [DOI] [PubMed] [Google Scholar]
- 87.Ye J, Garg A, Calhoun C, Feingold KR, Elias PM, Ghadially R. Alterations in cytokine regulation in aged epidermis: implications for permeability barrier homeostasis and inflammation. I. IL-1 gene family. Exp Dermatol 2002; 11:209–16; PMID:12102659; http://dx.doi.org/ 10.1034/j.1600-0625.2002.110303.x [DOI] [PubMed] [Google Scholar]
- 88.Hannigan GD, Grice EA. Microbial ecology of the skin in the era of metagenomics and molecular microbiology. Cold Spring Harb Perspect Med 2013; 3:a015362; PMID:24296350; http://dx.doi.org/ 10.1101/cshperspect.a015362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scharschmidt TC, List K, Grice EA, Szabo R, Renaud G, Lee CC, Wolfsberg TG, Bugge TH, Segre JA. Matriptase-deficient mice exhibit ichthyotic skin with a selective shift in skin microbiota. J Invest Dermatol 2009; 129:2435–42; PMID:19387477; http://dx.doi.org/ 10.1038/jid.2009.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schroder JM, Harder J. Antimicrobial skin peptides and proteins. Cell Mol Life Sci 2006; 63:469–86; PMID:16416029; http://dx.doi.org/ 10.1007/s00018-005-5364-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gallo RL, Nakatsuji T. Microbial symbiosis with the innate immune defense system of the skin. J Invest Dermatol 2011; 131:1974–80; PMID:21697881; http://dx.doi.org/ 10.1038/jid.2011.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, Hooper LV, Schmidt RR, von Aulock S, et al.. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med 2009; 15:1377–82; PMID:19966777; http://dx.doi.org/ 10.1038/nm.2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamasaki K, Gallo RL. Antimicrobial peptides in human skin disease. Eur J Dermatol 2008; 18:11–21; PMID:18086583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bauer B, Pang E, Holland C, Kessler M, Bartfeld S, Meyer TF. The Helicobacter pylori virulence effector CagA abrogates human beta-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe 2012; 11:576–86; PMID:22704618; http://dx.doi.org/ 10.1016/j.chom.2012.04.013 [DOI] [PubMed] [Google Scholar]
- 95.Shuyi Y, Feng W, Jing T, Hongzhang H, Haiyan W, Pingping M, Liwu Z, Zwahlen RA, Hongyu Y. Human beta-defensin-3 (hBD-3) upregulated by LPS via epidermal growth factor receptor (EGFR) signaling pathways to enhance lymphatic invasion of oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2011; 112:616–25; PMID:22035653; http://dx.doi.org/ 10.1016/j.tripleo.2011.02.053 [DOI] [PubMed] [Google Scholar]
- 96.Paruchuri V, Prasad A, McHugh K, Bhat HK, Polyak K, Ganju RK. S100A7-downregulation inhibits epidermal growth factor-induced signaling in breast cancer cells and blocks osteoclast formation. PLoS One 2008; 3:e1741; PMID:18320059; http://dx.doi.org/ 10.1371/journal.pone.0001741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stoll SW, Zhao X, Elder JT. EGF stimulates transcription of CaN19 (S100A2) in HaCaT keratinocytes. J Invest Dermatol 1998; 111:1092–7; PMID:9856822; http://dx.doi.org/ 10.1046/j.1523-1747.1998.00402.x [DOI] [PubMed] [Google Scholar]
- 98.Emberley ED, Niu Y, Curtis L, Troup S, Mandal SK, Myers JN, Gibson SB, Murphy LC, Watson PH. The S100A7-c-Jun activation domain binding protein 1 pathway enhances prosurvival pathways in breast cancer. Cancer Res 2005; 65:5696–702; PMID:15994944; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3927 [DOI] [PubMed] [Google Scholar]
- 99.Klingelhofer J, Moller HD, Sumer EU, Berg CH, Poulsen M, Kiryushko D, Soroka V, Ambartsumian N, Grigorian M, Lukanidin EM. Epidermal growth factor receptor ligands as new extracellular targets for the metastasis-promoting S100A4 protein. FEBS J 2009; 276:5936–48; PMID:19740107; http://dx.doi.org/ 10.1111/j.1742-4658.2009.07274.x [DOI] [PubMed] [Google Scholar]
- 100.Johnston A, Gudjonsson JE, Aphale A, Guzman AM, Stoll SW, Elder JT. EGFR and IL-1 signaling synergistically promote keratinocyte antimicrobial defenses in a differentiation-dependent manner. J Invest Dermatol 2011; 131:329–37; PMID:20962853; http://dx.doi.org/ 10.1038/jid.2010.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tanghetti EA. The role of inflammation in the pathology of acne. J Clin Aesthet Dermatol 2013; 6:27–35; PMID:24062871 [PMC free article] [PubMed] [Google Scholar]
- 102.Eilers RE Jr., Gandhi M, Patel JD, Mulcahy MF, Agulnik M, Hensing T, Lacouture ME. Dermatologic infections in cancer patients treated with epidermal growth factor receptor inhibitor therapy. J Natl Cancer Inst 2010; 102:47–53; PMID:20007525; http://dx.doi.org/ 10.1093/jnci/djp439 [DOI] [PubMed] [Google Scholar]
- 103.Gerber PA, Buhren BA, Steinhoff M, Homey B. Rosacea: the cytokine and chemokine network. J Invest Dermatol Symp Proc 2011; 15:40–7; PMID:22076326; http://dx.doi.org/ 10.1038/jidsymp.2011.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baldwin HE. Systemic therapy for rosacea. Skin Therapy Lett 2007; 12:1-5, 9; PMID:17393050 [PubMed] [Google Scholar]
- 105.Guy R, Ridden C, Kealey T. The improved organ maintenance of the human sebaceous gland: modeling in vitro the effects of epidermal growth factor, androgens, estrogens, 13-cis retinoic acid, and phenol red. J Invest Dermatol 1996; 106:454–60; PMID:8648176; http://dx.doi.org/ 10.1111/1523-1747.ep12343608 [DOI] [PubMed] [Google Scholar]
- 106.Philpott MP, Sanders D, Westgate GE, Kealey T. Human hair growth in vitro: a model for the study of hair follicle biology. J Dermatol Sci 1994; 7 Suppl:S55–72; PMID:7999676; http://dx.doi.org/ 10.1016/0923-1811(94)90036-1 [DOI] [PubMed] [Google Scholar]
- 107.Mak KK, Chan SY. Epidermal growth factor as a biologic switch in hair growth cycle. J Biol Chem 2003; 278:26120–6; PMID:12714603; http://dx.doi.org/ 10.1074/jbc.M212082200 [DOI] [PubMed] [Google Scholar]
- 108.Moore GP, Panaretto BA, Robertson D. Epidermal growth factor delays the development of the epidermis and hair follicles of mice during growth of the first coat. Anat Rec 1983; 205:47–55; PMID:6601466; http://dx.doi.org/ 10.1002/ar.1092050107 [DOI] [PubMed] [Google Scholar]
- 109.Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, Jenkins NA, Lee DC. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev 1994; 8:399–413; PMID:8125255; http://dx.doi.org/ 10.1101/gad.8.4.399 [DOI] [PubMed] [Google Scholar]
- 110.Murillas R, Larcher F, Conti CJ, Santos M, Ullrich A, Jorcano JL. Expression of a dominant negative mutant of epidermal growth factor receptor in the epidermis of transgenic mice elicits striking alterations in hair follicle development and skin structure. EMBO J 1995; 14:5216–23; PMID:7489711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hansen LA, Alexander N, Hogan ME, Sundberg JP, Dlugosz A, Threadgill DW, Magnuson T, Yuspa SH. Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am J Pathol 1997; 150:1959–75; PMID:9176390 [PMC free article] [PubMed] [Google Scholar]
- 112.Huang D, Liu X, Plymate SR, Idowu M, Grimes M, Best AM, McKinney JL, Ware JL. Proteomic identification of 14-3-3 sigma as a common component of the androgen receptor and the epidermal growth factor receptor signaling pathways of the human prostate epithelial cell line M12. Oncogene 2004; 23:6881–9; PMID:15300238; http://dx.doi.org/ 10.1038/sj.onc.1207788 [DOI] [PubMed] [Google Scholar]
- 113.Kalli KR, Bradley SV, Fuchshuber S, Conover CA. Estrogen receptor-positive human epithelial ovarian carcinoma cells respond to the antitumor drug suramin with increased proliferation: possible insight into ER and epidermal growth factor signaling interactions in ovarian cancer. Gynecol Oncol 2004; 94:705–12; PMID:15350362; http://dx.doi.org/ 10.1016/j.ygyno.2004.05.059 [DOI] [PubMed] [Google Scholar]
- 114.Egloff AM, Rothstein ME, Seethala R, Siegfried JM, Grandis JR, Stabile LP. Cross-talk between estrogen receptor and epidermal growth factor receptor in head and neck squamous cell carcinoma. Clin Cancer Res 2009; 15:6529–40; PMID:19825947; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-0862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Saif MW, Gnanaraj J. Erlotinib-induced trichomegaly in a male patient with pancreatic cancer. Cutan Ocul Toxicol 2010; 29:62–6; PMID:19954400; http://dx.doi.org/ 10.3109/15569520903440058 [DOI] [PubMed] [Google Scholar]
- 116.Rodeck U. Skin toxicity caused by EGFR antagonists-an autoinflammatory condition triggered by deregulated IL-1 signaling? J Cellul Physiol 2009; 218:32–4; PMID:18781585; http://dx.doi.org/ 10.1002/jcp.21585 [DOI] [PubMed] [Google Scholar]
- 117.Fletcher EV, Love-Homan L, Sobhakumari A, Feddersen CR, Koch AT, Goel A, Simons AL. EGFR inhibition induces proinflammatory cytokines via NOX4 in HNSCC. Mol Cancer Res 2013; 11:1574–84; PMID:24048704; http://dx.doi.org/ 10.1158/1541-7786.MCR-13-0187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Overdijk MB, Verploegen S, van den Brakel JH, Lammerts van Bueren JJ, Vink T, van de Winkel JG, Parren PW, Bleeker WK. Epidermal growth factor receptor (EGFR) antibody-induced antibody-dependent cellular cytotoxicity plays a prominent role in inhibiting tumorigenesis, even of tumor cells insensitive to EGFR signaling inhibition. J Immunol 2011; 187:3383–90; http://dx.doi.org/ 10.4049/jimmunol.1003926 [DOI] [PubMed] [Google Scholar]
- 119.Yang X, Zhang X, Mortenson ED, Radkevich-Brown O, Wang Y, Fu YX. Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Mol Ther 2013; 21:91–100; PMID:22990672; http://dx.doi.org/ 10.1038/mt.2012.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ocvirk J, Heeger S, McCloud P, Hofheinz RD. A review of the treatment options for skin rash induced by EGFR-targeted therapies: evidence from randomized clinical trials and a meta-analysis. Radiol Oncol 2013; 47:166–75; PMID:23801914; http://dx.doi.org/ 10.2478/raon-2013-0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shin JU, Park JH, Cho BC, Lee JH. Treatment of epidermal growth factor receptor inhibitor-induced acneiform eruption with topical recombinant human epidermal growth factor. Dermatology 2012; 225:135–40; PMID:23006507; http://dx.doi.org/ 10.1159/000342203 [DOI] [PubMed] [Google Scholar]
- 122.Perez-Soler R, Zou Y, Li T, Ling YH. The phosphatase inhibitor menadione (vitamin K3) protects cells from EGFR inhibition by erlotinib and cetuximab. Clin Cancer Res 2011; 17:6766–77; PMID:21914790; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0545 [DOI] [PubMed] [Google Scholar]
- 123.Tomkova H, Pospiskova M, Zabojnikova M, Kohoutek M, Serclova M, Gharibyar M, Sternbersky J. Phytomenadione pre-treatment in EGFR inhibitor-induced folliculitis. J Eur Acad Dermatol Venereol 2013; 27:514–9; PMID:22035385; http://dx.doi.org/ 10.1111/j.1468-3083.2011.04324.x [DOI] [PubMed] [Google Scholar]