

Abstract
Autophagy has opposite effects on hepatocarcinogenesis depending on whether it occurs before or after its onset. Autophagy prevents the initiation of hepatocarcinogenesis by suppressing oxidative stress and DNA damage. However, it also inhibits cell death and the expression of tumor suppressors to promote tumor progression once hepatocarcinogenesis has been initiated.
Keywords: autophagy, hepatocarcinogenesis, oxidative stress, tumor suppressors, tumor progression
Autophagy is important for maintaining cellular homeostasis and its dysfunction can cause a variety of diseases including neurodegeneration, aging, and autoimmune disorders. Autophagy is also associated with cancers, although its role in carcinogenesis is apparently complex as it was found to be protumorigenic in some studies but antitumorigenic in others.
Autophagy was originally thought to be antitumorigenic, as genes essential for autophagy, such as BECN1 (commonly known as the beclin-1 gene), UVRAG (UV radiation resistance-associated gene), ATG2B, ATG5, ATG9B, and ATG12, were frequently mutated or inactivated in a variety of cancer cells.1,2 The tumor suppressor role of autophagy had also been demonstrated using mouse models. It was shown that monoallelic deletion of Becn1 led to the development of tumor lesions in the lung, liver, and lymphatic systems;3 knockout of Atg4c increased the susceptibility of animals to carcinogens for the development of fibrosarcoma;4 and systemic mosaic knockout of Atg5 or liver-specific knockout of Atg7 led to the development of benign liver tumors.5 However, autophagy was also reported to be protumorigenic, as it was required to enhance the survival of tumor cells in the hypoxic regions of solid tumors and promoted tumorigenesis by maintaining oxidative metabolism and facilitating glycolysis.6,7 Moreover, inhibition of autophagy by the deletion of Rb1cc1 (RB1-inducible coiled coil protein 1, also known as FIP200) suppressed mammary tumorigenesis in mice.8
These reports on the conflicting roles of autophagy in carcinogenesis suggest that the effect of autophagy on carcinogenesis is complex and may be context dependent. To further understand the relationship between autophagy and carcinogenesis, we produced mice with hepatocyte-specific knockout of Atg5, a gene essential for autophagy, and longitudinally studied the role of autophagy in hepatocarcinogenesis.5 A fraction of these knockout mice developed liver tumors by 6 months of age and all of them developed liver tumors by 10 months. In contrast, no tumors were found in control mice by 12 months. By performing electron microscopy and a mitochondrial membrane potential assay, we noted the accumulation of defective mitochondria in the liver of Atg5-knockout mice. This accumulation of defective mitochondria was associated with increased levels of reactive oxygen species (ROS) and oxidative DNA damage, which provided an explanation for the increase in the incidence of liver tumors in these mice. The role of oxidative stress in the induction of liver tumors in Atg5-knockout mice was confirmed by treatment of these mice with the antioxidant N-acetylcysteine, which reduced the tumor incidence.
Interestingly, although Atg5-knockout mice developed liver tumors with a high frequency, histological analysis of their liver tumors revealed only focal nodular hyperplasia and benign adenomas with no malignant hepatocellular carcinoma (HCC). Treatment with the carcinogen diethylnitrosamine (DEN), which induced HCC with high frequencies in control mice, failed to induce HCC in these knockout mice. These results indicated that, in the absence of autophagy, benign liver tumors could not progress to become malignant HCC.
To understand why autophagy was required for the development of HCC, we analyzed the liver tumor tissues of Atg5-knockout mice and found an increase in apoptotic and necrotic cell death. We also analyzed the expression of tumor suppressor genes including Trp53 (commonly known as p53), Cdkn1a, Cdkn1b, Cdkn2a, Rb1, and Pten, and found that their expression levels were increased in the liver tumor tissues of Atg5-knockout mice. In contrast, the expression levels of these tumor suppressors were reduced in liver tumors isolated from control mice that had been treated with DEN. The increase in p53 expression in the liver of Atg5-knockout mice was associated with activation of the ATM (ataxia telangiectasia mutated)-CHEK2 (checkpoint kinase 2) signaling pathway and a reduced level of MDM2, an E3 ubiquitin protein-ligase that negatively regulates p53. The role of the ATM-CHEK2 pathway in the induction of p53 when autophagy was impaired was confirmed using the HepG2 human hepatoblastoma cell line. We found that suppression of ATG5 expression in HepG2 cells using small-hairpin RNA (shRNA) similarly led to activation of the ATM-CHEK2 pathway, suppression of MDM2 expression, and induction of p53 expression, and that this induction of p53 could be abolished by suppressing the expression of ATM with shRNA. By studying the tumorigenesis of HepG2 cells in nude mice, we confirmed that impairing autophagy could indeed suppress tumorigenesis and that this suppression could be partially reversed if the expression of p53 was also suppressed by shRNA. Two recent studies found that impairing autophagy induced the expression of p53 and suppressed the progression of KRAS (i.e., oncogenic RAS)-induced lung and pancreatic tumors in mice, and that this suppression of tumorigenesis could be partially restored if the expression of p53 was abolished.9,10 These findings, together with ours, indicate that autophagy is required for the progression of at least 3 different types of cancers, and that p53 plays a negative role in carcinogenesis when autophagy is impaired.
Our studies thus demonstrate that autophagy possesses both antitumorigenic and protumorigenic activities, depending on whether it occurs before or after the onset of hepatocarcinogenesis (Fig. 1). Autophagy is important for removing damaged mitochondria and if autophagy is impaired dysfunctional mitochondria will accumulate to cause oxidative stress, DNA damage, and the initiation of hepatocarcinogenesis. However, after the initiation of hepatocarcinogenesis, autophagy is required to suppress the expression of tumor suppressors, alleviate metabolic stress,6 and reduce apoptotic and necrotic cell death, thus promoting the malignant transformation of benign tumors into HCC.
Figure 1.
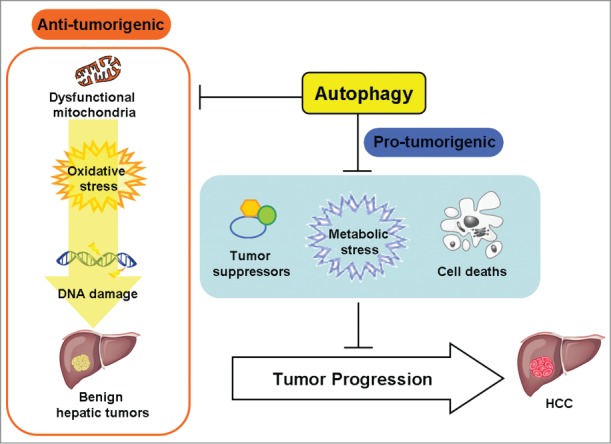
Dual effect of autophagy on hepatocarcinogenesis. Autophagy can be antitumorigenic, as it removes damaged mitochondria to suppress oxidative stress and DNA damage thus preventing the initiation of hepatocarcinogenesis. However, autophagy is also protumorigenic once hepatocarcinogenesis has been initiated, as it inhibits the expression of tumor suppressors, alleviates metabolic stress, and reduces cell death, thus allowing the development of hepatocellular carcinoma (HCC).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by research grants DK094652, DK100257 and CA177337 from the National Institutes of Health.
References
- 1.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672–6; PMID:10604474; http://dx.doi.org/ 10.1038/45257 [DOI] [PubMed] [Google Scholar]
- 2.Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, Ahn CH, Yoo NJ, Lee SH. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 2009; 217:702–6; PMID:19197948; http://dx.doi.org/ 10.1002/path.2509 [DOI] [PubMed] [Google Scholar]
- 3.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al.. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809–20; PMID:14638851; http://dx.doi.org/ 10.1172/JCI20039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 2007; 282:18573–83; PMID:17442669; http://dx.doi.org/ 10.1074/jbc.M701194200 [DOI] [PubMed] [Google Scholar]
- 5.Tian Y, Kuo C, Sir D, Wang L, Govindarajan S, Petrovic LM, Ou JJ. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ 2014: (in press); PMID:25526090; http://dx.doi.org/ 10.1038/cdd.2014.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathew R, Karantza-Wadsworth V, White E. Assessing metabolic stress and autophagy status in epithelial tumors. Methods Enzymol 2009; 453:53–81; PMID:19216902; http://dx.doi.org/ 10.1016/S0076-6879(08)04004-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al.. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460–70; PMID:21317241; http://dx.doi.org/ 10.1101/gad.2016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25:1510–27; PMID:21764854; http://dx.doi.org/ 10.1101/gad.2051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al.. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27:1447–61; PMID:23824538; http://dx.doi.org/ 10.1101/gad.219642.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al.. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013; 504:296–300; PMID:24305049; http://dx.doi.org/ 10.1038/nature12865 [DOI] [PubMed] [Google Scholar]