

Abstract
Common fragile sites (CFSs) tend to break upon replication stress and have been suggested to be “hot spots” for genomic instability. Recent evidence, however, implies that in the wake of DNA damage, WW domain-containing oxidoreductase (WWOX, the gene product of the FRA16D fragile site), associates with ataxia telangiectasia-mutated (ATM) and regulates its activation to maintain genomic integrity.
Genomic instability is a major risk factor for cancer development and progression.1 Multiple tumor suppressors are involved in maintaining the integrity of chromosome structure. The gene encoding WW domain-containing oxidoreductase (WWOX) is a tumor suppressor gene that spans the common fragile site (CFS) FRA16D.2 CFSs are chromosomal regions that tend to break in response to replication stress and loss of CFS-encoded genes is frequently observed in human cancer.3 The high susceptibility of CFSs to defects in DNA replication, impaired DNA damage response (DDR), and oxidative stress led to the notion that the genes spanning these sites are early sensors of DNA damage.4 Defects in CFS-associated genes such as WWOX and FHIT result in an increased risk of cancer progression, suggesting that rather than being inert sensors of DNA damage these genes play an active role as tumor suppressors.2-4 However, the mechanism by which the CFS-encoded genes contribute to tumor suppression remains largely unknown. Since genomic instability is a main driving force of carcinogenesis, we hypothesized that the CFS gene WWOX might play a direct role in the DNA damage response and genome stability. Indeed, our findings identified WWOX as a regulator of DNA damage repair and showed that WWOX is essential for proper activation of the key DNA damage checkpoint kinase ataxia telangiectasia-mutated (ATM).5 Therefore, we conclude that WWOX acts as an active driver, rather than a passive passenger, in the DNA damage response to prevent genome instability and tumorigenesis.
Previously, it was shown that loss of WWOX expression is associated with poor prognosis in numerous cancers.6 Wwox knockout (KO) mice exhibit post-natal lethality and die by 4 weeks of age, indicating the essential function of WWOX.7,8 WWOX encodes a 46-kDa protein that contains two N-terminal WW domains and a central short-chain dehydrogenase/reductase domain. WWOX interacts with proline-tyrosine motif-containing proteins via its WW1 domain and regulates their localization and transcriptional function.9
In our recent paper by Abu-Odeh et al.,5 we address the role of WWOX in DNA damage signaling and DNA repair. We found that WWOX expression was induced within minutes in response to induction of double-strand breaks (DSBs). The induction of WWOX early after DNA damage was observed in different human cancer cell lines and in mouse embryonic fibroblasts (MEFs), indicating that this is a common phenomenon rather than a cell-type specific event. Subcellular fractionation showed that in response to DSBs ITCH, an E3 ubiquitin ligase, mediates K63-linked polyubiquitination of WWOX at lysine (K) 274, facilitating its stabilization and nuclear accumulation.5 Importantly, depletion or genetic knockout of WWOX resulted in elevated levels of DSBs and reduced ATM signaling upon DNA damage.5 Reconstitution experiments in which WWOX was introduced demonstrated that wild-type WWOX, but not WWOX mutants harboring a defect in the WW1 domain (WWOX-WFPA) or in K274 (K274R), rescued the ATM activating function, as shown by phosphorylation of the ATM substrate KRAB-associated protein-1 (KAP1). Furthermore, WWOX mediates efficient DNA damage repair. In fact, our findings demonstrated that cells expressing wild-type WWOX, but not mutated WWOX, displayed more efficient microhomology-dependent repair and homologous recombination.5 Together, these experiments demonstrate the critical role of WWOX in DDR.
How does WWOX affect DDR? Our findings revealed that WWOX physically and functionally associates with ATM and mediates its activation. Coimmunoprecipitation studies showed that wild-type WWOX is required for its interactions with the Ser1981-phosphorylated active isoform of ATM (p-ATM) and with the ATM substrate p-KAP1 upon the induction of DSBs. Notably, in the absence of WWOX significantly less pATM was found at the DNA damage foci that mark the sites of DSBs. In addition, impaired function of the ATM checkpoint was demonstrated by reduced activation of mediator of DNA damage checkpoint protein 1 (MDC1) and decreased recruitment of pCHK2 to the DNA breaks, two DNA damage mediators that are activated in an ATM-dependent fashion.5 A possible function of WWOX in the ATM pathway is its contribution to ATM activation. ATM activation in response to DNA damage involves dissociation of inactive dimers into active monomers. Our results revealed that WWOX is required for proper monomerization of ATM, and hence its activation.
In summary, our study provides evidence that in response to DNA damage a subtle amount of WWOX accumulates in the cell nucleus where it interacts with pATM and mediates its proper activation, thus ensuring proper DDR and antagonizing genomic instability (Fig. 1).
Figure 1.
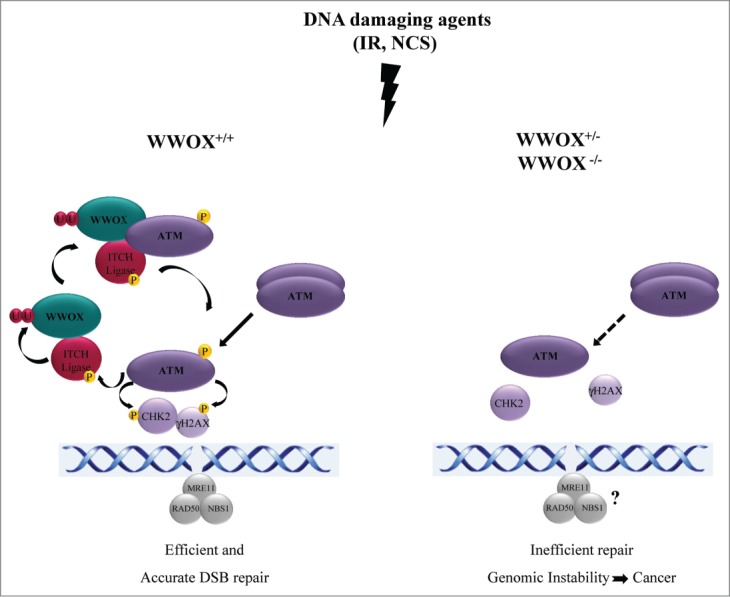
Involvement of WW domain-containing oxidoreductase (WWOX) in DNA double-strand break (DSBs) repair. (A) Induction of DSBs by ionizing radiation (IR) or neocarzinostatin (NCS) leads to activation of ataxia telangiectasia-mutated (ATM) through its monomerization and phosphorylation. Activated ATM phosphorylates and positively regulates the ligase activity of ITCH as well as other substrates including histone variant H2AX, checkpoint kinase 2 (CHK2), and tumor protein p53 (TP53, best known as p53). ITCH ubiquitinates WWOX on Lys274 and thereby promotes its accumulation in the nucleus. Nuclear WWOX physically interacts with ATM and facilitates its monomerization and activation through a positive feed-forward loop. (B) WWOX depletion (hemizygous mutation) or loss (homozygous mutation) results in reduced activation of ATM, decreased phosphorylation of H2AX and CHK2, and defects in the recruitment of p-ATM, γ-H2AX, and p-CHK2 to DNA damage sites. Overall, loss of WWOX results in delayed activation of the DNA damage checkpoint kinase ATM and impaired DNA repair.
Analysis of Wwox-deficient mice led to the identification of pleiotropic functions of WWOX in homeostasis and tumor suppression.6-8 For example, recent studies associated WWOX loss with aerobic glycolysis and impaired cellular metabolism, therefore it is reasonable to assume that various WWOX functions collectively lead to tumor suppression.10 Our recent study suggests that the fragile site gene product WWOX behaves as a genome ‘caretaker’ and that its loss leads to genome instability and cancer development.2,5 This novel role of WWOX in the activation of DSB repair raises various questions: What is the exact function of WWOX in DSB repair? Does WWOX participate in repair of other types of DNA damage beside DSBs? Does WWOX also function in endogenous (metabolically-caused) DNA damage repair? In addition, this study raises questions concerning the genomic instability in WWOX-related cancers: Is there a predominant mutational signature and/or specific rearrangement pattern? Does WWOX deficiency lead to other human diseases associated with impaired DDR? And finally, do other CFS gene products have similar functions in guarding the genome and would combined deletion/alteration of these genes have a more pronounced effect on tumor promotion? Future studies aimed at answering these questions may provide insights into how WWOX, and other CFS gene products, prevent genomic instability and why such essential tumor suppressor genes were maintained within CFSs during evolution.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11:220–8; PMID:20177397; http://dx.doi.org/ 10.1038/nrm2858 [DOI] [PubMed] [Google Scholar]
- 2.Aqeilan RI, Abu-Remaileh M, Abu-Odeh M. The common fragile site FRA16D gene product WWOX: roles in tumor suppression and genomic stability. Cell Mol Life Sci 2014; 71:4589–99; PMID:25245215; http://dx.doi.org/ 10.1007/s00018-014-1724-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao G, Smith DI. Very large common fragile site genes and their potential role in cancer development. Cell Mol Life Sci 2014; 71:4601–15; PMID:25300511; http://dx.doi.org/ 10.1007/s00018-014-1753-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Georgakilas AG, Tsantoulis P, Kotsinas A, Michalopoulos I, Townsend P, Gorgoulis VG. Are common fragile sites merely structural domains or highly organized “functional” units susceptible to oncogenic stress? Cell Mol Life Sci 2014; 71:4519–44; PMID:25238782; http://dx.doi.org/ 10.1007/s00018-014-1717-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abu-Odeh M, Salah Z, Herbel C, Hofmann TG, Aqeilan RI. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc Natl Acad Sci U S A 2014; 111:E4716–4725; PMID:25331887; http://dx.doi.org/ 10.1073/pnas.1409252111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardenswartz A, Aqeilan RI. WW domain-containing oxidoreductase's role in myriad cancers: Clinical significance and future implications. Exp Biol Med 2014; 239:253–63; PMID:24510053; http://dx.doi.org/ 10.1177/1535370213519213 [DOI] [PubMed] [Google Scholar]
- 7.Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y, Hagan JP, Zanesi N, Kaou M, Stein GS, et al.. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci U S A 2007; 104:3949–54; PMID:17360458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aqeilan RI, Hassan MQ, de Bruin A, Hagan JP, Volinia S, Palumbo T, Hussain S, Lee SH, Gaur T, Stein GS, Lian JB, et al.. The WWOX tumor suppressor is essential for post-natal survival and normal bone metabolism. J Biol Chem 2008; 283:21629–39; PMID:18487609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abu-Odeh M, Bar-Mag T, Huang H, Kim T, Salah Z, Abdeen SK, Sudol M, Reichmann D, Sidhu S, Kim PM, et al.. Characterizing WW domain interactions of tumor suppressor WWOX reveals Its association with multiprotein networks. J Biol Chem 2014; 289:8865–80; PMID:24550385; http://dx.doi.org/ 10.1074/jbc.M113.506790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abu-Remaileh M, Aqeilan RI. Tumor suppressor WWOX regulates glucose metabolism via HIF1alpha modulation. Cell Death Differ 2014; 21:1805–14; PMID:25012504; http://dx.doi.org/ 10.1038/cdd.2014.95 [DOI] [PMC free article] [PubMed] [Google Scholar]