

Abstract
Acute myeloid leukemia (AML) is the most common type of leukemia in adults. Development of resistance to chemotherapeutic agents is a major hurdle in the effective treatment of patients with AML. The quinazolinone derivative erastin was originally identified in a screen for small molecules that exhibit synthetic lethality with expression of the RAS oncogene. This lethality was subsequently shown to occur by induction of a novel form of cell death termed ferroptosis. In this study we demonstrate that erastin enhances the sensitivity of AML cells to chemotherapeutic agents in an RAS-independent manner. Erastin dose-dependently induced mixed types of cell death associated with ferroptosis, apoptosis, necroptosis, and autophagy in HL-60 cells (AML, NRAS_Q61L), but not Jurkat (acute T-cell leukemia, RAS wild type), THP-1 (AML, NRAS_G12D), K562 (chronic myelogenous leukemia, RAS wild type), or NB-4 (acute promyelocytic leukemia M3, KRAS_A18D) cells. Treatment with ferrostatin-1 (a potent ferroptosis inhibitor) or necrostatin-1 (a potent necroptosis inhibitor), but not with Z-VAD-FMK (a general caspase inhibitor) or chloroquine (a potent autophagy inhibitor), prevented erastin-induced growth inhibition in HL-60 cells. Moreover, inhibition of c-JUN N-terminal kinase and p38, but not of extracellular signal-regulated kinase activation, induced resistance to erastin in HL-60 cells. Importantly, low-dose erastin significantly enhanced the anticancer activity of 2 first-line chemotherapeutic drugs (cytarabine/ara-C and doxorubicin/adriamycin) in HL-60 cells. Collectively, the induction of ferroptosis and necroptosis contributed to erastin-induced growth inhibition and overcame drug resistance in AML cells.
Keywords: AML, autophagy, erastin, ferroptosis, necroptosis
Introduction
The most common type of leukemia in adults is acute myeloid leukemia (AML), which is characterized by the rapid growth of abnormal lineage-specific hematopoietic precursor cells that do not differentiate into functional granulocytes or monocytes during hematopoiesis in the bone marrow. Treatment of AML includes remission induction and post-remission therapy (e.g., chemotherapy or allogeneic stem cell transplantation).1 Combination treatment with cytosine arabinoside (cytarabine/ara-C) and an anthracycline has been the cornerstone of AML treatment since its introduction in the 1960s2 although doxorubicin/adriamycin was used then instead of daunorubicin/cerubidin, which is now the more common anthracycline for human AML therapy. Approximately 70–80% of patients aged less that 60 years achieve complete remission, but most ultimately relapse. In addition, all-trans–retinoic acid induces complete remission in acute promyelocytic leukemia (APL), which is the M3 subtype of AML according to the French–American–British classification.3
Poor outcomes in AML patients are related to drug resistance and relapse. Many mechanisms confer drug resistance, such as impaired drug export transporters (e.g., permeability-glycoprotein), altered drug target sites, abnormal pharmacokinetics, and resistance to programmed cell death (PCD).4,5 PCD is regulated by an intricate mechanism and dysfunction of PCD is closely related to anticancer therapy.6 Apoptosis mediated by caspases is a well-studied type of PCD.7 In addition to apoptosis, several novel types of PCD such as necroptosis and ferroptosis have been recognized to be involved in cancer cell death induced by chemotherapeutic agents. Necroptosis is a programmed form of necrosis that is involved in the inflammatory response,8,9 whereas ferroptosis is a form of non-apoptotic cell death that depends on iron signaling.10 Autophagy, or programmed cell survival (PCS), is an alternative mechanism of resistance to AML therapies.11 Understanding the molecular and cellular basis of PCD and PCS is critical in the development of effective cancer treatments.
Members of the RAS family of small GTPases, including HRAS, NRAS, and KRAS, are mutated in 33% of cancers, stimulating an intensive effort to develop RAS inhibitors for cancer therapy.12 The quinazolinone derivative erastin was originally identified in a screen for small molecules that are synthetically lethal with expression of the RAS oncogene.13 In cancer cells, erastin induces an iron-dependent cell death that lacks the classic characteristics of apoptosis, necrosis, necroptosis, and autophagy.10 In addition to solid cancer, RAS mutations are frequently activated in patients with blood cancers, including those with leukemia.14 RAS mutations occur in 25% of patients with AML and enhance sensitivity to chemotherapeutic agents, including cytarabine.15 The anticancer activity of erastin in leukemia cells and whether the RAS mutation influences its action remain to be elucidated.
We found that erastin selectively enhances the sensitivity of non-APL AML cells (e.g., HL-60 cells) to chemotherapeutic agents (e.g., cytarabine and doxorubicin) in an RAS-independent manner. This was accompanied by cooperative induction of ferroptosis and necroptosis in a selective mitogen-activated protein kinase (MAPK)-dependent manner. Our results suggest an alternative set of mechanisms distinct from those of ferroptosis alone underlying the action of erastin in leukemia cells.
Results
Erastin induces growth inhibition in HL-60 cells
To determine whether erastin induces growth inhibition in leukemia cells, 5 individual human leukemia cell lines were treated with erastin for 24 h and cell viability was assayed using the Cell Counting Kit-8 (CCK-8). Interestingly, erastin dose-dependently induced growth inhibition in HL-60 cells (non-APL AML, NRAS_Q61L), but not in Jurkat (acute T cell leukemia, RAS wild type), THP-1 (acute monocytic leukemia, NRAS_G12D), K562 (chronic myelogenous leukemia, RAS wild type), or NB-4 cells (APL, KRAS_A18D) (Fig. 1A). Thus RAS mutation status did not appear to have a major impact on the growth inhibition response of leukemia cells exposed to erastin (Fig. 1B and C). These findings suggest that erastin selectively induces growth inhibition in leukemia cells in an RAS-independent manner.
Figure 1.
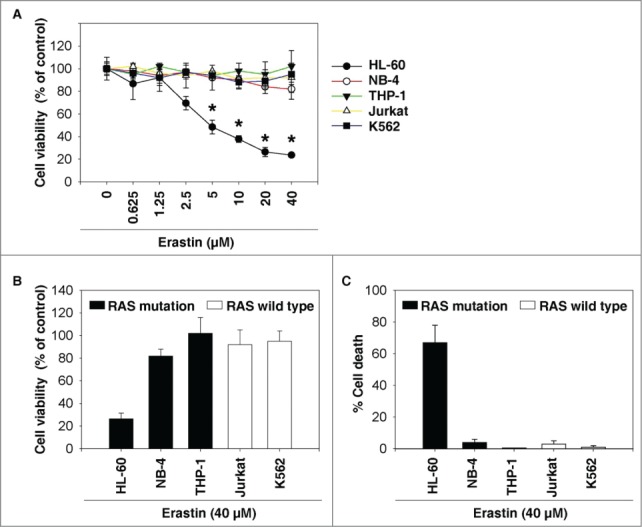
Erastin inhibits growth of HL-60 cells. (A) The indicated leukemia cells were treated with erastin at the indicated doses for 24 h and cell viability was assayed using a CCK-8 kit (n=3, *P<0.05 versus untreated group). (B, C) The indicated leukemia cells were treated with erastin (40 μM) for 24 h. Cell viability was assayed using the CCK-8 kit (B) and the dead cells were counted using a trypan blue exclusion assay (C).
Erastin induces mixed types of cell death in HL-60 cells
Previous studies indicate that erastin induces ferroptosis, but not other types of PCD, in cancer cells derived from several solid tumor types.10,16,17 To investigate whether erastin has similar effects on HL-60 cells, we assayed protein markers for ferroptosis (glutathione peroxidase 4 [GPX4]), apoptosis (cleaved-poly ADP ribose polymerase [PARP] and cleaved-caspase 3), autophagy (microtubule-associated protein 1 light chain 3 [LC3] and p62), necrosis (high mobility group protein B1 [HMGB1], and lactate dehydrogenase [LDH]) using western blotting techniques. GPX4 is a negative regulator of ferroptosis.16 Erastin inhibited the expression of GPX4 in HL-60 cells (Fig. 2A) and the human osteosarcoma U2OS cell line (a positive control cell line that responds by ferroptosis) (Fig. 2A). Surprisingly, erastin also induced cleaved-PARP, cleaved-caspase 3, LC3-II expression, and p62 degradation in whole-cell extracts and HMGB1/LDH release in culture supernatants from HL-60, but not U2OS, cells (Fig. 2A). These findings suggest that erastin induces a mixed type of cell death in HL-60 cells. In contrast, this response to erastin treatment was not observed in Jurkat cells (Fig. 2A). Intracellular chelatable iron was determined using the fluorescent indicator phen green SK, fluorescence of which is quenched by iron. The proportion of phen green SK-positive cells in HL-60 cells decreased after treatment with erastin (Fig. 2B), suggesting that iron may be involved in erastin-induced cell death.
Figure 2.
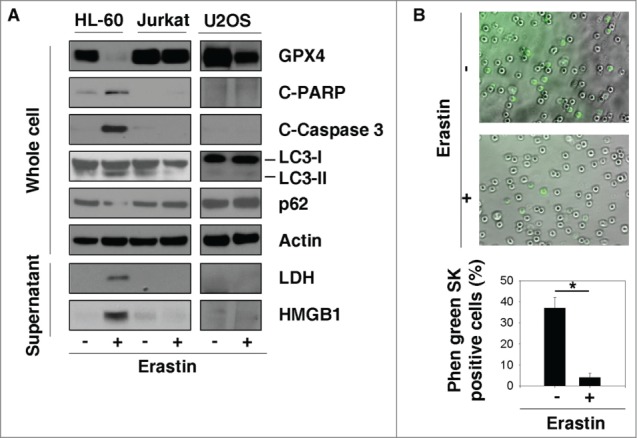
Erastin induces mixed types of cell death in HL-60 cells. (A) HL-60 and Jurkat cells were treated with erastin (5 μM) for 24 h and subjected to western blot analysis of the indicated proteins in whole cell extracts or supernatant. (B) Intracellular chelatable iron in HL-60 cells treated with or without erastin (5 μM, 24 h) was determined using the fluorescent indicator phen green SK (green). A non-fluorescent differential interference contrast image and a fluorescent image were collected simultaneously. Phen green SK-positive cells were quantified in a minimum of ten 20 × microscopic fields and expressed as a percentage (*, P ˂ 0.05).
Ferroptosis and necroptosis contribute to erastin-induced growth inhibition in HL-60 cells
To characterize the role of cell death in erastin-induced growth inhibition, we treated HL-60 cells with erastin in the absence or presence of several potential cell death inhibitors. Treatment with deferoxamine (an iron-chelating agent), ferrostatin-1 (a potent inhibitor of ferroptosis), or necrostatin-1 (a potent inhibitor of necroptosis), but not with Z-VAD-FMK (a general caspase inhibitor) or chloroquine (a potent inhibitor of autophagy), prevented erastin-induced growth inhibition in HL-60 cells (Fig. 3A). In contrast, Z-VAD-FMK and chloroquine inhibited HL-60 cell death induced by staurosporine (apoptotic inducer) and HBSS (autophagic inducer) respectively (Fig. 3B). Moreover, knockdown of receptor-interacting protein 3 (RIP3, a regulator of necroptosis) by specific shRNA inhibited erastin-induced growth inhibition in HL-60 cells, but not in U2OS cells (Fig. 3C). These findings indicate that ferroptosis and necroptosis, but not apoptosis and autophagy, contribute to erastin-induced growth inhibition in HL-60 cells.
Figure 3.

Ferroptosis and necroptosis contribute to erastin-induced growth inhibition in HL-60 cells. (A) HL-60 cells were treated with erastin (5 μM) with or without the indicated inhibitors for 24 h and cell viability was assayed (n=3, *P < 0.05 versus erastin treatment group). (B) HL-60 cells were treated with the apoptotic inducer staurosporine (1 μM) and the autophagic inducer HBSS (100%) with or without the indicated inhibitors for 24 h and cell viability was assayed (n=3, *P < 0.05). (C) The indicated RIP3 knockdown cells were treated with erastin (5 μM) for 24 h and cell viability was assayed (n=3, *P < 0.05).
Inhibition of JNK and p38 activation induces resistance to erastin in HL-60 cells
Next, we investigated the signaling regulatory pathways involved in erastin-induced growth inhibition. Three major MAPKs—c-JUN N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase (ERK)—regulate PCD and the immune response.18,19 Erastin promoted phosphorylation of JNK and p38, but not of ERK, in HL-60 cells (Fig. 4A). SP600125 (an inhibitor of JNK phosphorylation) and SB202190 (an inhibitor of p38 activation) significantly decreased the cytotoxicity induced by erastin (Fig. 4B). In contrast, PD98059 (an inhibitor of the ERK upstream activators MAPK kinase [MKK] 1 and MKK2) had no influence on erastin-induced cell death (Fig. 4B). SP600125 and SB202190, but not PD98059, inhibited erastin-induced HMGB1 release (Fig. 4C). Moreover, knockdown of p38α (a major isoform of p38) by specific shRNA inhibited erastin-induced growth supression in HL-60 cells (Fig. 4D). Collectively, these findings suggest that JNK and p38 cooperatively participate in cell death induced by erastin in HL-60 cells.
Figure 4.
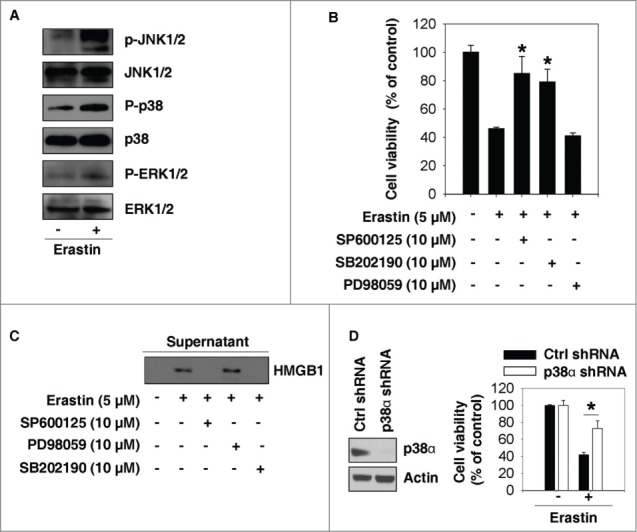
Inhibition of JNK and p38 activation induces resistance to erastin in HL-60 cells. (A) HL-60 cells were treated with erastin (5 μM) for 24 h and subjected to western blot analysis of the indicated proteins in whole cell extracts. (B, C) HL-60 cells were treated with erastin (5 μM) with or without the indicated inhibitors for 24 h and cell viability was assayed (n=3, *P < 0.05 versus erastin treatment group) (B). In parallel, HMGB1 in the supernatant was analyzed using western blotting (C). (D) The indicated p38α knockdown HL-60 cells were treated with erastin (5 μM) for 24 h and cell viability was assayed (n=3, *P < 0.05).
Erastin enhances anticancer activity of cytarabine and doxorubicin in HL-60 cells
We also investigated whether erastin enhances the anticancer activity of 2 first-line chemotherapy drugs, cytarabine and doxorubicin, in AML cells. Erastin at a concentration of 1.25 μm did not significantly affect growth inhibition in HL-60 cells. However, this low cytotoxic dose of erastin remarkably enhanced the anticancer activity of both cytarabine (Fig. 5A) and doxorubicin (Fig. 5B). Treatment with ferrostatin-1 and necrostatin-1 prevented combination therapy-induced growth inhibition in HL-60 cells (Fig. 5A and B). In contrast, Z-VAD-FMK and chloroquine had no effect on growth inhibition (Fig. 5A and B). Thus, these findings indicate that low-dose erastin enhances the anticancer activity of cytarabine and doxorubicin in non-APL AML cells, in part by induction of ferroptosis and necroptosis.
Figure 5.
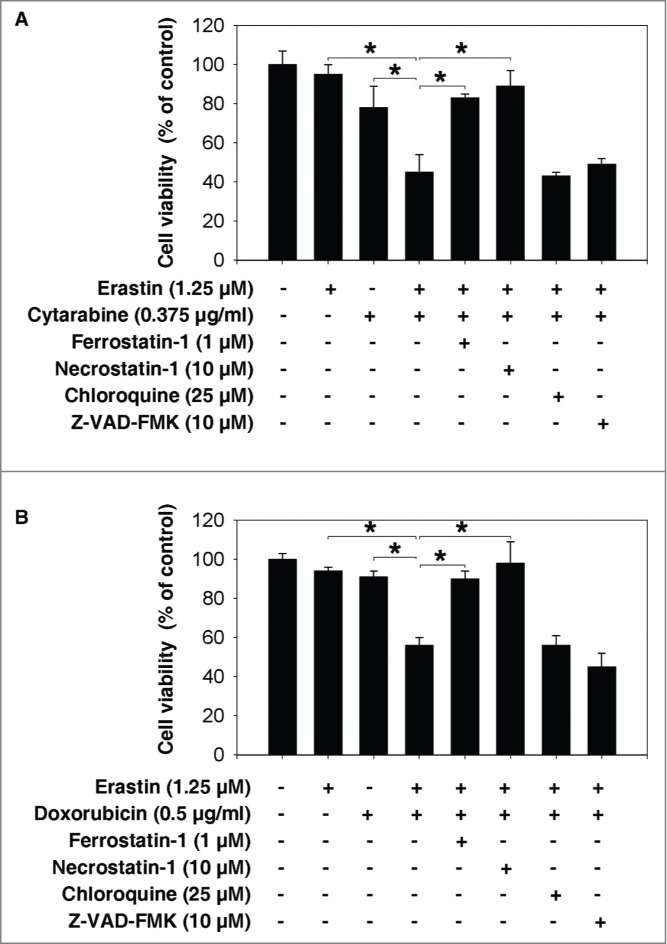
Erastin enhances the anticancer activity of cytarabine and doxorubicin in HL-60 cells. HL-60 cells were treated with cytarabine (A) and doxorubicin (B) combined with erastin in the absence or presence of the indicated inhibitors for 24 h and cell viability was assayed (n = 3, *P < 0.05).
Discussion
In this study, we demonstrated that erastin elicits not only ferroptosis but also other forms of PCD such as necroptosis, which contributes to growth inhibition in HL-60 cells. The molecular mechanisms underlying the cooperative induction of ferroptosis and necroptosis are involved in the activation of JNK and p38 signaling. In addition to exhibiting high antitumor activity as a single agent at a high concentration, a low concentration of erastin enhances the sensitivity of HL-60 cells to chemotherapeutic agents. Our findings may therefore provide novel treatment options for patients with non-APL AML.
Previous studies indicate that erastin is an RAS-selective lethal compound that kills hyperproliferating cancer cells through the induction of ferroptosis.13,17,20 We demonstrated that the anticancer activity of erastin in leukemia cells is RAS-independent. Ferroptosis, as a novel mechanism of PCD, is morphologically, biochemically, and genetically distinct from other types of cell death including apoptosis, necrosis, and autophagy; for example, the classic features of apoptosis, including caspase activation and DNA fragmentation, are not observed in ferroptosis.10 In addition, characteristic morphological features of necrosis, such as disruption of the cellular membrane, are not observed in ferroptosis.10 In contrast, increased iron-mediated production of lipid reactive oxygen species is an essential feature of ferroptosis processes.10 Several positive (e.g., voltage-dependent anion channels17,21) and negative (e.g., GPX4,16,22 heat-shock protein β-1 [HSPB1],23 and cystine/glutamate antiporters10,24) regulators have recently been demonstrated to regulate ferroptotic cancer cell death, although their action and specificity remain unclear. Our findings indicate that erastin induces a mixed type of cell death, including both ferroptosis and necroptosis, in HL-60 cells. This process can be blocked by deferoxamine, ferrostatin-1, and necrostatin-1, but not by Z-VAD-FMK or chloroquine. In addition to their effect on cancer cells, erastin and other ferroptosis inducers also promote necroptosis in normal kidney cells.22,25 This process can be reversed by novel second- and third-generation ferrostatins with higher plasma stability in vivo.25 Collectively, these findings suggest that the genetic background of individual cell types may influence the type of erastin-mediated cancer cell death.
MAPKs are activated by a variety of environmental stressors and regulate multiple cell processes ranging from cell survival to cell death.18,19 Although the signal transduction mechanism associated with ferroptosis remains largely unknown, our findings indicate that JNK and p38, but not the ERK MAPK pathway, are responsible for erastin-induced cell death in HL-60 cells. The ERK-dependent signaling pathway is, however, required for RAS-dependent ferroptosis in solid cancer cells.17 Thus, these findings suggest that activation of individual MAPK pathways may influence the anticancer activity of erastin in solid cancer and leukemia cells. In future studies, exploiting the specificity of MAPK substrates may prove a viable alternative to distinguish various erastin-induced types of cell death.
Several treatment options are currently available for AML patients. The best treatment recommendation depends on the subtype of AML and cytogenetic characteristics of the leukemic cells.26,27 Although a number of chemotherapeutic agents are effective against AML, the goal of treatment is to eliminate malignant cells and reduce toxicity to normal cells. Our findings indicate that erastin enhances the anticancer activity of cytarabine and doxorubicin, 2 first-line chemotherapy drugs for the remission induction of non-APL AML, by enhancing both necroptosis and ferroptosis. These mechanisms of cell death have only been described relatively recently; much work remains to enable means of targeting these pathways for cancer treatment. For example, we have recently demonstrated that HSPB1, a small heat-shock protein that is important in regulating autophagy28 and apparently important in AML,29 is also critically important for ferroptosis.23 These observations may provide insights allowing the development of novel combination therapies to overcome resistance in AML patients. In addition, GPX4 expression prevents against ferroptosis after T-cell activation,30 suggesting that targeting the ferroptotic pathway may contribute to immunotherapy for human diseases, including autoimmune diseases and cancer.31
Materials and Methods
Antibodies and reagents
Antibodies to GPX4 (#ab125066) and actin (Ab3280) were obtained from Abcam (Cambridge, MA, USA). Antibodies to ERK (#4695), P-ERK (#9101), JNK (#9252), P-JNK (#9251), p38 (#9212), p38α (#9218), P-P38 (#9215), LDH (#3582), cleaved-PARP (#9541), and cleaved-caspase 3 (#9661) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies to HMGB1 (#H00003146-M08) and LC3 (#NB600-1384) came from Novus (Littleton, CO, USA). The antibody to p62 (#Sc-28359) was obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and the antibody to RIP3 (#31513) was from Sigma (St. Louis, MO, USA). Z-VAD-FMK (#V116), deferoxamine (#D9533), necrostatin-1 (#N9037), chloroquine (#C6628), SP600125 (#S-5567), PD 98059 (#P215), SB-202190 (#S8307), cytarabine (#C1768), and doxorubicin (#D1515) were obtained from Sigma. Ferrostatin-1 (#S7243) and erastin (#S7242) were obtained from Selleck Chemicals (Houston, TX, USA).
Cell culture
HL-60 (#CCL-240), Jurkat (#TIB-152), THP-1 (#TIB-202), K562 (#CCL-243) and U2OS (#HTB-96) cells were obtained from ATCC (Manassas, VA, USA). NB-4 was a gift from Dr. Michel Lanotte (INSERM U-301, Paris, France).32 These cells were grown in Dulbecco's Modified Eagle's Medium, RPMI-1640 Medium, or McCoy's 5a Medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 100 U/ml penicillin and streptomycin.
Cell viability analysis
Cell viability was evaluated using a CCK-8 Kit (#CK04, Dojindo Molecular Technologies, Tokyo, Japan) according to the manufacturer's instructions. Briefly, leukemic cells (2 × 104/well) were seeded in a 96-well plate and treated with different drugs at various concentrations for the indicated times. After addition of 10 μl CCK-8 solution to each well, cells were incubated at 37°C for another 3 h and the absorbance was determined at 450 nm using a microplate reader. Trypan blue exclusion testing of cell viability was also conducted in parallel.
RNAi
Human RIP3-shRNA (SHCLNG-NM_006871_ TRCN 0000234968; sequence: CCGGTCGTAAACTCGAAGGCGATATCTCGAGATATCGCCTTCGAGTTTACGATTTTTG), human p38α-shRNA (SHCLNG-NM_001315_ TRCN0000000511; sequence: CCGGCCATGAGGCAAGAAACTATATCTCGAGATATAGTTTCTTGCCTCATGGTTTTT), and control shRNA (SHC001) were obtained from Sigma. Transfections were performed with Lipofectamine™ 3000 (#L3000015, Invitrogen) or the Neon® Transfection System (Invitrogen) according to the manufacturer's instructions.
Western blot analysis
Proteins in whole cell extracts or cell media were analyzed by western blotting as described previously.28 Briefly, after extraction the proteins were resolved by electrophoresis on a 4–12% Criterion™ XT Bis-Tris Gel (Bio-Rad, Hercules, CA, USA), transferred to a nitrocellulose membrane (pore size 0.45 μm), and incubated with the appropriate primary antibody (1:500-1:1,000 dilution). After incubation with peroxidase-conjugated secondary antibody (1:2,000), antibody binding was visualized using enhanced chemiluminescence reagents (Pierce, Rockford, IL, USA, #32106) followed by exposure to standard X-ray films.
Iron assay
Intracellular chelatable iron was determined using the fluorescent indicator phen green SK (#P-14313, Life Technologies, Grand Island, NY, USA), the fluorescence of which is quenched by iron. Imaging was performed using the EVOS® FL Auto Cell Imaging System (Life Technologies).
Statistical analysis
Data are expressed as means ± SD of 3 independent experiments and were evaluated using an ANOVA LSD test. P values <0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for critical reading of the manuscript.
Funding
This work was supported by the National Institutes of Health (NIH) (R01CA160417 to DT), a grant from the National Natural Sciences Foundation of China (81270616 to Y.Y; 31171328 and 81370648 to LC), and Core Support from the University of Pittsburgh Cancer Institute (P30CA047904).
References
- 1. Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia 2014; 29(4):760-769; PMID:25142817 [DOI] [PubMed] [Google Scholar]
- 2. Yates JW, Wallace HJ, Jr., Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother Rep 1973; 57:485-8; PMID:4586956 [PubMed] [Google Scholar]
- 3. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, Shepherd L, Willman C, Bloomfield CD, Rowe JM, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med 1997; 337:1021-8; PMID:9321529; http://dx.doi.org/ 10.1056/NEJM199710093371501 [DOI] [PubMed] [Google Scholar]
- 4. Sakamoto KM, Grant S, Saleiro D, Crispino JD, Hijiya N, Giles F, Platanias L, Eklund EA. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol Genet Metab 2014; 114(3):397-402; PMID:25533111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Loze MT, et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy 2011; 7:112-4; PMID:21068541; http://dx.doi.org/ 10.4161/auto.7.1.14005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 2015; 22:58-73; PMID:25236395; http://dx.doi.org/ 10.1038/cdd.2014.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35:495-516; PMID:17562483; http://dx.doi.org/ 10.1080/01926230701320337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature 2015; 517:311-20; PMID:25592536; http://dx.doi.org/ 10.1038/nature14191 [DOI] [PubMed] [Google Scholar]
- 9. Linkermann A, Green DR. Necroptosis. N Engl J Med 2014; 370:455-65; PMID:24476434; http://dx.doi.org/ 10.1056/NEJMra1310050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149:1060-72; PMID:22632970; http://dx.doi.org/ 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sehgal AR, Konig H, Johnson DE, Tang D, Amaravadi RK, Boyiadzis M, Lotze MT. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia 2015; 29:517-25; PMID:25541151; http://dx.doi.org/ 10.1038/leu.2014.349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 2014; 13:828-51; PMID:25323927; http://dx.doi.org/ 10.1038/nrd4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003; 3:285-96; PMID:12676586; http://dx.doi.org/ 10.1016/S1535-6108(03)00050-3 [DOI] [PubMed] [Google Scholar]
- 14. Reuter CW, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood 2000; 96:1655-69; PMID:10961860 [PubMed] [Google Scholar]
- 15. Neubauer A, Maharry K, Mrozek K, Thiede C, Marcucci G, Paschka P, Mayer RJ, Larson RA, Liu ET, Bloomfield CD. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: a Cancer and Leukemia Group B study. J Clin Oncol 2008; 26:4603-9; PMID:18559876; http://dx.doi.org/ 10.1200/JCO.2007.14.0418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156:317-31; PMID:24439385; http://dx.doi.org/ 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007; 447:864-8; PMID:17568748; http://dx.doi.org/ 10.1038/nature05859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004; 23:2838-49; PMID:15077147; http://dx.doi.org/ 10.1038/sj.onc.1207556 [DOI] [PubMed] [Google Scholar]
- 19. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 2013; 13:679-92; PMID:23954936; http://dx.doi.org/ 10.1038/nri3495 [DOI] [PubMed] [Google Scholar]
- 20. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008; 15:234-45; PMID:18355723; http://dx.doi.org/ 10.1016/j.chembiol.2008.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM, Bezrukov SM, Rostovtseva TK, Lemasters JJ. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem 2013; 288:11920-9; PMID:23471966; http://dx.doi.org/ 10.1074/jbc.M112.433847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014; 16:1180-91; PMID:25402683; http://dx.doi.org/ 10.1038/ncb3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, Wang H, Cao L, Tang D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015; [Epub ahead of print]; PMID:25728673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014; 3:e02523; PMID:24844246; http://dx.doi.org/ 10.7554/eLife.02523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 2014; 111:16836-41; PMID:25385600; http://dx.doi.org/ 10.1073/pnas.1415518111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chung SS. Genetic mutations in acute myeloid leukemia that influence clinical decisions. Curr Opin Hematol 2014; 21:87-94; PMID:24445361; http://dx.doi.org/ 10.1097/MOH.0000000000000024 [DOI] [PubMed] [Google Scholar]
- 27. Khasawneh MK, Abdel-Wahab O. Recent discoveries in molecular characterization of acute myeloid leukemia. Curr Hematol Malig Rep 2014; 9:93-9; PMID:24609756; http://dx.doi.org/ 10.1007/s11899-014-0200-y [DOI] [PubMed] [Google Scholar]
- 28. Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 2011; 13:701-11; PMID:21641551; http://dx.doi.org/ 10.1016/j.cmet.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang L, Cao L, Yang M, Tang D, Kang R, Min X, Zhu S, Yu Y. Hsp27: a novel therapeutic target for pediatric M4/M5 acute myeloid leukemia. Oncol Rep 2013; 29:1459-66; PMID:23404246 [DOI] [PubMed] [Google Scholar]
- 30. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 2015; 212:555-68; PMID:25824823; http://dx.doi.org/ 10.1084/jem.20140857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol 2014; 14:759-67; PMID:25324125; http://dx.doi.org/ 10.1038/nri3743 [DOI] [PubMed] [Google Scholar]
- 32. Lanotte M, Martin-Thouvenin V, Najman S, Balerini P, Valensi F, Berger R. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 1991; 77:1080-6; PMID:1995093 [PubMed] [Google Scholar]