

Abstract
PIK3R1 (encoding the p85α subunit of phosphatidylinositol 3-kinase) is the 11th most frequently mutated gene across tumors. We recently reported neomorphic p85α mutants that induce signaling cascades not predicted by the canonical functions of p85α, suggesting the need to functionally annotate specific mutations in cancer genes for effective genome-informed personalized therapy.
Keywords: MAPK, mutation, PIK3R1, p85a, PI3K, neomorph
Class 1A phosphoinositide 3-kinase (PI3K) is a heterodimeric lipid kinase composed of a p85 regulatory subunit and p110 catalytic subunit that activates protein kinase B (AKT) and other downstream effectors. Aberrations of components of the PI3K pathway are collectively the most common activating events in cancers. PIK3R1 (encoding p85α, which has been proposed to be the critical regulator of p110) is also frequently mutated; indeed, PIK3R1 is the 11th most commonly mutated gene across cancer lineages in The Cancer Genome Atlas database.1 Previously characterized PIK3R1 mutations exclusively target PI3K pathway activation. We and others have demonstrated several hypomorphic (decrease in function) PIK3R1 mutations that activate the PI3K pathway by relieving the inhibition of p110α or by destabilizing phosphatase and tensin homolog (PTEN) protein.2,3 In a recent study, we described an unexpected neomorphic (gain of novel function) role for a subset of PIK3R1 mutations derived from cancer patients.4 This subset of mutations, including R348* and neighboring truncation mutations, represents the most common recurrent PIK3R1 mutations, accounting for approximately 10% of all PIK3R1 mutations in endometrial and colon cancers and therefore justifying the exploration of approaches able to benefit patients with these aberrations.
Our first step leading to the neomorph concept was an unbiased drug screen with a library of 150 compounds targeting major signaling pathways. The data revealed that the p85α neomorphs rendered cells sensitive to mitogen-activated protein kinase kinase (MEK) and c-Jun N-terminal protein kinases (JNK) inhibitors. This was completely unexpected because p85α does not normally impinge on the mitogen-activated protein kinase (MAPK) pathway. In support of these data, wild-type p85α and other known oncogenic p85α mutants had no effect on sensitivity to the MEK and JNK inhibitors. We next undertook functional proteomics analysis of the mutants through a reverse phase protein array that covers proteins of cancer-relevant signaling pathways. In complete accord with the drug sensitivity data, the neomorphs, but not wild-type or other p85α mutants, induced phosphorylation of extracellular signal regulated kinase (ERK) and JNK, indicating that activation of the pathway underlies drug sensitivity. Subsequent mechanistic studies revealed that the neomorphs resulted in selective activation of components of the ERK and JNK signaling cascades.
The activation of MAPK pathways by the neomorphs is independent of the conventional role of p85α in PI3K signaling. First, the activation was insensitive to PI3K inhibitors. Second, the truncation mutation produces a protein that cannot bind to p110, and thus the neomorphic activity is unlikely to be mediated through p110. This p110-independency and the lack of intrinsic kinase activity of p85α led to the major question of how the neomorphs activate MAPK pathways, and particularly nuclear JNK. Strikingly, we discovered that, in contrast to wild-type p85α, the neomorphs were present in the nucleus where they acted as a scaffold to tether JNK signaling components in close proximity thereby facilitating JNK activation. This distinct neomorphic characteristic compared with wild-type p85α and the other p85α mutants is probably attributable to the p85α protein domains present that lead to differential binding partners (Fig. 1). The neomorphs were translocated into the nucleus by binding to nuclear transport chaperones including the small GTPases cell division cycle 42 (Cdc42) and Ras-related C3 botulinum toxin substrate 1 (Rac1) through the intact p85α breakpoint-cluster region homology (BH) domain. Although wild-type p85α and some other p85α mutants contain the BH domain, and indeed bind these nuclear chaperones, their nuclear import is prohibited, possibly because of physical linkage to cytoskeletal and membrane proteins mediated by the src homology 2 (SH2) domains.
Figure 1.
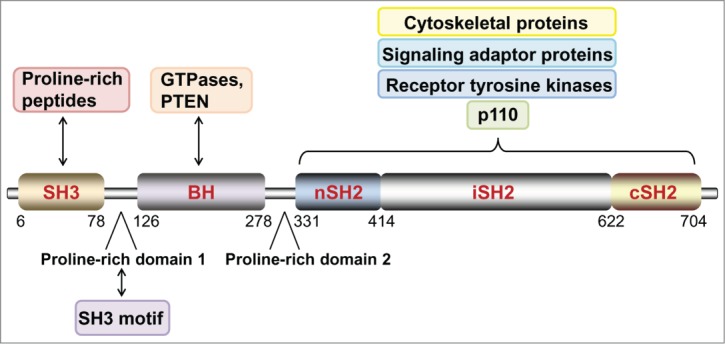
Structural organization and major binding partners of wild-type p85α. The function and localization of wild-type p85α and its truncation variants may be determined by its binding partners. SH3, src homology 3; BH, breakpoint-cluster region homology; nSH2, N-terminal src homology 2; iSH2, inter-Src homology 2; cSH2, C-terminal src homology 2.
p85α that is not bound to p110 can undergo homodimerization that may alter the conformation of the protein, thus exposing motifs that bind specific sets of proteins. We have demonstrated that the wild-type p85α homodimer binds and stabilizes PTEN.2 This binding was disrupted in a truncated mutant E160* lacking the BH and SH2 domains, which does not translocate into the nucleus or alter the catalytic activity of p110. Therefore, homodimerization resulting from p85α truncation proteins of different sizes may alter the binding partners. Given that the neomorphs do not bind p110, whether the neomorphic effects are mediated by a homodimer or by a heterodimeric form of the mutant with wild-type p85α remains to be investigated.
Our observations that xenograft tumors expressing neomorphs were susceptible to MAPK inhibitors and that endometrial cancer patients bearing neomorphs had high levels of ERK and JNK phosphorylation may provide a rationale for therapeutic targeting of these mutant tumors. These mutations could potentially be biomarkers of responsiveness to inhibitors targeting the ERK and JNK pathways. Since the neomorphs also activate the PI3K pathway4 and activation of the MAPK pathway by Ras mutants can mediate resistance to therapy targeting the PI3K pathway,5 it may be necessary to treat patients with these neomorphs using a combination of MEK and PI3K pathway inhibitors. A combinatorial approach of MEK and PI3K pathway inhibition is being assessed in several clinical trials for colon cancer and endometrial cancer patients.
More importantly, our identification and characterization of neomorphs constitutes an important conceptual advance suggesting that targeted therapies may need to be conditioned on the specific aberration in the cancer gene rather than on the cancer gene alone. To date, neomorphic mutations have also been reported in IDH1, IDH2, and TP53.6,7 Functional characterization of patient-specific mutations may be needed to fulfill the promise of personalized cancer therapy and, importantly, prevent untoward consequences of targeted therapy. This is especially true for genes that are linked to specific targeted therapies. Identification and characterization of neomorphs is particularly challenging because available approaches to predict the function of mutations, including protein structure modeling, may have limited robustness for neomorphic actions. The experimental pipeline established in this study is a proof-of-concept that can be implemented in other functional genomics studies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Supported by NCI 2P50 CA098258–06, NCI U01 CA168394, Stand Up to Cancer/AACR Dream Team Translational Cancer Research Grant SU2C-AACR-DT0209, TCGA GDAC Grant (NIH/NCI U24 CA143883) to GBM; MDACC Uterine SPORE Career Development Award (NCI P50CA098258) to LWT.
References
- 1. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. . The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-404; PMID:22588877; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, et al. . High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov 2011; 1:170-185; PMID:21984976; http://dx.doi.org/ 10.1158/2159-8290.CD-11-0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, et al. . Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 2009; 16:463-474; PMID:19962665; http://dx.doi.org/ 10.1016/j.ccr.2009.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N, Mitra S, Ju Z, Yu Q, Liang H, et al. . Naturally Occurring Neomorphic PIK3R1 Mutations Activate the MAPK Pathway, Dictating Therapeutic Response to MAPK Pathway Inhibitors. Cancer Cell 2014; 26:479-494; PMID:25284480; http://dx.doi.org/ 10.1016/j.ccell.2014.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ihle NT, Lemos R, Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res 2009; 69:143-150; PMID:19117997; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-6656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. . The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17:225-234; PMID:20171147; http://dx.doi.org/ 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Y, Prives C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 2007; 26:2220-2225; PMID:17401431; http://dx.doi.org/ 10.1038/sj.onc.1210311 [DOI] [PubMed] [Google Scholar]