

Abstract
Our current knowledge of the molecular mechanisms regulating the signaling pathways leading to cell survival, cell death, and inflammation has shed light on the tight mutual interplays between these processes. Moreover, the fact that both apoptosis and necrosis can be molecularly controlled has greatly increased our interest in the roles that these types of cell death play in the control of general processes such as development, homeostasis, and inflammation. In this review, we provide a brief update on the different cell death modalities and describe in more detail the intracellular crosstalk between survival, apoptotic, necroptotic, and inflammatory pathways that are activated downstream of death receptors. An important concept is that the different cell death processes modulate each other by mutual inhibitory mechanisms, serve as alternative back-up death routes in the case of a defect in the first-line cell death response, and are controlled by multiple feedback loops. We conclude by discussing future perspectives and challenges in the field of cell death and inflammation research.
Keywords: apoptosis, necroptosis, crosstalk, tumor necrosis factor, Fas, RIPK1, RIPK3, FLIP
Abbreviations
- TNF
tumor necrosis factor
- DAMPs
damage associated molecular patterns
- PAMPs
pathogen associated molecular patterns
- DR
death receptors
- TLR
Toll-like receptors
- NLR
NOD-like receptors
- MOMP
mitochondrial outer membrane permeabilization
- IRI
ischemia reperfusion injury
- RIPK1
receptor interacting protein kinase 1
- RIPK3
receptor interacting protein kinase 3
- MLKL
mixed lineage kinase domain-like
- NSA
necrosulfonamide
- 4HBD
four-helical bundle domain
- MPT
mitochondrial permeability transition
- CypD
cyclophilin D
- ROS
reactive oxygen species
- PARP1
Poly(ADP-ribose) (PAR) polymerase 1
- DD
death domain
- DISC
death inducing signaling complex
- LUBAC
linear ubiquitin assembly complex
- c-FLIP
FLICE-like inhibitory proteins
- MEFs
mouse embryonic fibroblasts
- SMAC
second mitochondria-derived activator of caspase
- CHX
cycloheximide
Inflammation and cell death: a forced marriage? The oldest references to inflammation are found on Egyptian papyri dating from around 1650 BCE. More than 2000 years ago, Celsus (25 BC – c. 50 AD, Rome) described the 4 cardinal signs of inflammation as calor (heat), rubor (redness), tumor (swelling), and dolor (pain). Although initially considered a passive pathological consequence of injury characterized by these 4 hallmarks, inflammation has gradually emerged as an active process that initiates at the site of infection or injury and functions to control infection and promote tissue repair. Galen of Pergamon (approximately 200 AD) initiated this change of concept by proposing that the generation of pus is a sign of healing. Rudolf Virchow (1821–1902, Prussia), founder of the Cell Theory (“Omnis cellula e cellula”), cellular pathology, and even social medicine, later referred to such tissue injury as “parenchymatous inflammation” and introduced the idea that injured tissue can be traced to pathological changes intrinsic to the cells themselves. In 1858, he introduced the notion of cell death as a potential basis for pathology through the opposing processes of “necrobiosis,” a physiological process of spontaneous wearing out of living parts from the body, and “necrosis,” an accidental process. Virchow's necrobiosis–necrosis dichotomy resembles to some extent the current apoptosis–necrosis classification.1 Together with the evolving ideas and molecular insights in inflammation came a shift in our understanding of the molecular interplay between cell death and inflammation at the site of tissue injury. Although cell death in the course of inflammation was initially thought to be a manifestation of tissue damage, it was later recognized as a mechanism of eliminating pathogens and regulating inflammation by exposing cellular components or their unique derivatives that attract and alter the functions of other cells.2
The molecular interplay between inflammation and cell death is an emerging field of research that is crucial for the understanding of organismal homeostasis and how these processes contribute to a growing list of inflammatory and degenerative pathologies. The current notion that both apoptotic and necrotic cell death is molecularly controlled by defined signaling mechanisms has increased our interest in the regulatory roles of cell death in general processes such as development, homeostasis, and inflammation (reviewed elsewhere3). This increased understanding is the foundation that allows us to take the first steps toward therapeutic validation to resolve an excess or lack of cell death in certain pathologies.4 In this review, we briefly introduce the different cell death modalities and highlight their morphological, biochemical, and functional properties. We describe in more detail the different molecular routes to different cell death modalities, focusing on the intracellular crosstalk between them. We will present evidence that different cell death processes often modulate each other by mutual inhibitory mechanisms, back up each other in the case of defective first-line cell death responses, and are controlled by multiple feedback loops, similar to many other cellular processes. This redundancy in cell death signaling nicely illustrates the plasticity of nature. On one hand this creates multiple options to induce cell death, but on the other hand it makes therapeutic targeting and clinical translation very challenging. We conclude by discussing some current perspectives and challenges in the field of cell death and inflammation research.
A Snapshot of Cell Death Prototypes
Although the first morphological descriptions of cellular demise date back to the mid-nineteenth century, the categorical terms “programmed cell death” or “apoptosis” were only formulated in the late 1960s and early 1970s.5,6 The first morphological classification of cell death was proposed based on observations in rat embryos exposed to toxicants.7 Today, these cell death modes are referred to as apoptosis (type I), autophagic cell death (type II), and necrosis (type III).8 During the 1980s and 1990s apoptosis was found to be genetically controlled, as was elegantly demonstrated in Caenorhabiditis elegans (reviewed by Lettre et al.9). Textbooks soon contrasted apoptosis with necrosis, the latter being considered a purely accidental and passive cell death. However, the unregulated nature of necrosis was soon questioned when it was discovered that tumor necrosis factor (TNF) was able to elicit either the “classic” features of apoptosis or a “balloon-like” morphology without nuclear disintegration, depending on the cell type.10 Since then, accumulating evidence has paved the way to the concept of regulated necrosis.11 Before going into the molecular details, it is important to note that there are multiple forms of regulated necrosis (Table 1).3 Below is a brief description of the best-characterized cell death types.8
Table 1.
Schematic overview of the best-characterized genetically regulated cell death prototypes: apoptosis versus regulated necrosis
| Apoptosis | Regulated necrosis | |||||||
|---|---|---|---|---|---|---|---|---|
| Morphology | Pseudopod retraction Rounding up Decreased cellular volume Chromatin condensation Nuclear fragmentation Blebbing of the plasma membrane Shedding of apoptotic bodies | Increasingly translucent cytoplasm Often swelling of organelles | Chromatin decondensation | |||||
| Increased cell volume (oncosis) culminating in disruption of the plasma membrane | ||||||||
| Sub category | Intrinsic apoptosis | Extrinsic apoptosis | Necroptosis | Ferroptosis | MPT-mediated RN | Parthanatos | Pyroptosis | (N)Etosis |
| Key molecules | BID, BAX/BAD CASP9 APAF1 Cytc | RIPK1a, RIPK3 FADD CASP8 CASP2? | RIPK1b RIPK3 MLKL | GPX4 | CypD | PARP1 | CASP1 CASP11 | NOX |
| Death execution | CASP3 CASP7 | TRPM7 Pores | GSH decrease Fe | [Ca2+]↑ | [NAD+]↓ [ATP]↓ | ROS↑ | ||
| Lipid peroxidation, energetic catastrophe, LMP | ||||||||
| Synthetic Inhibitor | zVAD-fmk qVD-oph | Nec-1 Nec-1s | Fer-1 | Sf(A) CsA | 3-AB PJ-34 | VX-740 VX-765 | DPI GKT | |
| Physiology | Morphology during embryonic development Control of cell number during homeostasis Pathogen defense | Competes with DR-induced apoptosis | Glu toxicity Kills cancer cells | Transplantation Thrombosis | DNA damage | Professional inflammatory form of RN | Extracellular trap formation | |
| Pathogen defense Causative link to several pathologies | ||||||||
Under cIAP-depleting conditions or TAK1 inhibition;
requirement dependent on the trigger.
Abbreviations: CASP, Caspase; MPT, mitochondrial permeability transition; BID, Bcl-2 interacting domain; BAX, Bcl-2 associated X protein; BAD, Bcl-2 antagonist of cell death; RIPK, receptor interacting protein kinase; MLKL, mixed lineage kinase domain-like; GPX4, gluthatione peroxidase 4; CypD, cyclophilin D; PARP1, Poly(ADP-ribose) (PAR) polymerase 1; NOX, NADPH oxidases; TRPM7, transient receptor potential melastatin 7; GSH, glutathione; Fe, iron; Ca, calcium; NAD, nicotinamide adenine dinucleotide; ATP, adenosine triphosphate; ROS, reactive oxygen species; LMP, lysosomal membrane permeabilisation; zVAD-fmk, benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone; qVD-oph, quinolyl-valyl-O-methylaspartyl-(-2, 6-difluorophenoxy)-methyl ketone; Nec, necrostatin; Fer, ferrostatin; Sf(A), sanglifehrin A; CsA, cyclosporin A; 3-AB, 3-aminobenzamide; PJ-34, PARP inhibitor; VX-740 and VX765, caspase-1 inhibitor; DPI, diphenyleneiodonium; GKT, NOX inhibitor; Glu, glutamate; RN, regulated necrosis; c-IAP, cellular inhibitor of apoptosis; TAK1, TGF-β-activated kinase 1
Apoptosis and necroptosis
Apoptosis is a caspase-dependent cell death modality. The proteolytic activation of caspases in apoptotic cells drives cell rounding, retraction of pseudopodes, reduction of cellular volume (pyknosis), chromatin condensation, nuclear fragmentation (karyorrhexis), and plasma membrane blebbing. Even though there is minimal ultrastructural modification of cytoplasmic organelles, the cell is systematically dismantled into membrane-wrapped vesicles (apoptotic bodies) that in vivo are rapidly engulfed by resident phagocytes, thus preventing exposure of intracellular components to the immune system. In the absence of swift clearance by phagocytes, apoptotic bodies undergo secondary necrosis during which the membrane integrity of the dead cell corpses is lost. Two distinct pathways regulate apoptosis: the extrinsic and intrinsic pathways. The extrinsic pathway refers to apoptotic cell death triggered by extracellular stress signals such as damage or pathogen-associated molecular patterns (DAMPs or PAMPs) or cytokines that are sensed and propagated primarily by a subset of transmembrane receptors of the tumor necrosis factor (TNF)-superfamily called death receptors (DR), Toll-like receptors (TLR) or NOD-like receptors (NLR). Apoptosis induced by the intrinsic pathway occurs in response to a wide range of intracellular damage or stress signals including cytokine withdrawal, DNA damage, oxidative stress, endoplasmic reticulum (ER) stress, and cytosolic Ca2+ overload that converge on mitochondrial outer membrane permeabilization (MOMP) and the release of mitochondrial factors such as cytochrome C, Smac/DIABLO, or Omi/HtrA2.12,13 In a cell type-specific fashion, the extrinsic machinery also engages the intrinsic pathway via processing of BID therefore allowing a proapoptotic amplification loop.
Regulated necrosis manifests with similar morphological features as passive necrosis but death is executed in a cell autonomous fashion via distinct biochemical processes leading to cellular rounding followed by swelling (oncosis), cytoplasmic granulation, and plasma membrane rupture. Loss of membrane integrity provokes an inflammatory response by exposing leaking cellular contents to the immune system. Regulated necrosis, characterized by oncosis and plasma membrane permeabilization, includes multiple cell death sub-classes such as necroptosis, parthanatos, ferroptosis, (n)etosis, pyroptosis, and ischemia reperfusion injury (IRI)-mediated necrosis. Each type of regulated necrosis highlights particular biochemical features, yet it is still unclear whether shared pathways or converging pathways underline the common morphological features of these multiple forms of cell death.3 Necroptosis is currently the best-characterized form of regulated necrosis and is mediated by the conserted action of receptor interacting protein kinase 1 (RIPK1) and RIPK3, and mixed lineage kinase domain-like (MLKL) in response to death receptors, Toll- and NOD-like receptors, T-cell receptor, genotoxic stress, and viruses.14-16 The chemical inhibitors necrostatin-1,17 GSK'843/'87218 and necrosulfonamide (NSA)19 block necroptosis by respectively inhibiting m/hRIPK1, hRIPK3, and hMLKL. Of interest is the recently identified action mechanism of MLKL. The four-helical bundle domain (4HBD) in the N-terminal region of MLKL is required and sufficient to induce oligomerization, bind phosphatidylinositol phosphates, permeabilize membranes, and induce cell death.20-22 It seems that this membrane permeabilizing activity of 4HBD is controlled by a masking helix H6, which interacts not only with the helix H2-H5 interface but also with helix H4 that contains Cys86 (the target of NSA). Hence it is likely that inhibition by NSA in the liposome leakage assays that is mediated by covalent modification of C86 disrupts intra- or intermolecular interactions that are critical for the formation of pores within the membranes.21
Other forms of regulated necrosis beyond necroptosis
Mitochondrial permeability transition (MPT)-regulated necrosis contributes to ischemia reperfusion injury and acute organ failure. The mitochondrial matrix protein cyclophilin D (CypD) controls the MPT pore, and opening of the pore leads to translocation of NAD+ from the mitochondrial matrix to the cytosol.3
Ferroptosis involves the production of iron-dependent reactive oxygen species (ROS). Blockade of a cysteine/glutamate antiporter that supplies cells with oxidized cysteine suppresses glutathione biosynthesis. Lipid peroxidation and cell death occur as a consequence of a decrease in glutathione levels and depletion of GPX-4.23 A chemical inhibitor of ferroptosis is Ferrostatin-1, but its molecular target remains unclear.24
The process of parthanatos depends on poly(ADP-ribose) (PAR) polymerase 1 (PARP1) activation in response to DNA breakage, ROS, alkylating agents, the Ca2+ signaling pathway, or post-translational modifications such as phosphorylation, acetylation, or ADP ribosylation.25 The PARylation of proteins is thought to deplete cells of NAD+ (and consequently ATP), resulting in regulated necrosis.3
Pyroptosis is proposed to be a form of regulated necrosis that typically occurs in specialized immune cells to combat infection. Pyroptosis is a caspase-1– or caspase-11–dependent cell death modality that occurs downstream of inflammasome activation and is typically accompanied by maturation and release of the cytokines IL-1β and IL-18 as well as other activators of the immune system.26 Pyroptosis is associated with cellular swelling and plasma membrane permeabilization, but does not involve the necroptosis regulators. This highly inflammatory form of cell death is a defense mechanism against microbial infection and occurs primarily in infected macrophages and in the dying T cells of patients with AIDS.27,28
Similar to pyroptosis, NETosis/ETosis also occurs in specialized immune cells (such as neutrophils, other granulocytes, and macrophages) to combat infections. This form of regulated necrosis is characterized by chromatin decondensation and the release of (neutrophil) extracellular traps.29 These postmortem traps are composed of DNA, chromatin, and histones, and enable immune cells to immobilize and kill bacteria.30 It should be noted that the release of extracellular traps has also been reported in the absence of cell death.31
Mutual Interplay Between Cell Death and Gene Activation
The understanding of death receptor (DR)-mediated signaling pathways has been at the forefront of many fundamental developments in cell biology, including the molecular interplay between apoptosis, regulated necrosis, and inflammatory signaling. There are 6 human DRs in the TNF superfamily: TNFR1, CD95 (also known as FAS or APO-1), TRAILR1 (also known as DR4), TRAILR2 (also known as APO-2, TRICK, DR5, or KILLER), DR3 (also known as TRAMP or APO-3), and DR6 (reviewed by Walczak et al.32). These receptors induce death via their common cytoplasmic death domain (DD). Soon after the discovery of TNF it became clear that the most prominent outcome of engagement of TNFR1 by this ligand was not cell death, but instead the induction of cytokines and chemokines. In contrast, induction of cell death is the dominant consequence of FASL or TRAIL sensing by their respective receptors. In the case of TNFR1 signaling, the current model is that sensing of TNF by TNFR1 induces the assembly of a primary receptor-bound complex that triggers activation of signaling pathways leading to gene induction. In a subsequent stage, assembly of a secondary TNFR1-unbound cytosolic complex induces cell death.32 In the case of FAS and TRAILR1/2, the opposite situation is observed. Although the receptor-bound primary complex, the death inducing signaling complex (DISC), triggers cell death, the secondary cytosolic complex regulates gene activation (Fig. 1). The physiologic relevance of this sequential signaling is fine-tuning of the cellular response and provision of an alternative or back-up response by the secondary cytosolic complex in case the default pathway activated by the receptor-associated complex fails to dominate. Pathogens or (epi)genetic factors can interfere with gene activation or cell death induction. Thus, this sequential signaling probably evolved as a host defense strategy to contend with pathogens or malicious conditions that may perturb either pathway.
Figure 1.

Signaling of death receptors for cell death and survival. (A)The primary response of the inflammatory death receptors TNFR1 and DR3 is complex I-mediated gene activation, which is required for a proper inflammatory response. (B) The primary response of the prototype death receptors Fas and TRAILR is induction of cell death. Upon interference with this primary response, for example by pathogens, (epi)genetic factors, or chemical inhibitors, a secondary response prevails. In the case of TNFR1 and DR3 this secondary signal is complex II-mediated cell death (C), whereas gene induction is typically observed as a secondary signal downstream of Fas and TRAILRs (D). Abbreviations: DR3, death receptor 3; TNFR1, tumor necrosis factor receptor 1.
TNFR1-induced gene activation versus cell death
TNF is a master regulator of inflammation and cell death. Consequently, signaling downstream of TNFR1 is the best characterized among all DRs. Nuclear factor kappa B (NF-κB)–mediated gene activation by TNFR1 requires the formation of a receptor-associated TNFR signaling complex called complex I.33 In brief, cross-linking of TNFR1 by TNF promotes recruitment of TNF receptor–associated death domain (TRADD) and RIPK1 to the death domain (DD) of the receptor (Fig. 2). Subsequently, TRADD recruits TNF receptor–associated factor 2 (TRAF2), which in turn provides the platform for cellular inhibitor of apoptosis (cIAP)1/2 binding. cIAP1/2 then conjugate components of complex I with ubiquitin chains generated from various types of ubiquitin linkages. These modifications allow docking of linear ubiquitin assembly complex (LUBAC), which adds linear ubiquitin chains to NEMO and possibly other components of the complex such as RIPK1. The ubiquitin chains generated by cIAP1/2 and LUBAC enable recruitment and exact positioning of both the IKKα/IKKβ/NEMO and TAB/TAK-complexes (reviewed by Estornes et al.34). The concerted action of the latter 2 complexes results in the activation of MAPK and NF-κB signaling pathways (often referred to as the early signaling phase) that induce expression of genes encoding prosurvival and proliferative molecules, cytokines and chemokines, barrier molecules, protease inhibitors, and antioxidants (reviewed by Ben-Neriah et al.35). Termination of TNF-induced NF-κB activation requires dismantling of the ubiquitin network of complex I. The deubiquitylating enzyme USP21 shuts down the early signaling phase,36 whereas the A20 ubiquitin (Ub)-editing complex, including TAXBP1/Itch/RNF11 proteins, terminates NF-κB activation at later phases.37,38 In marked contrast to previous theories, the deubiquitylase activity of A20 was recently demonstrated to be dispensable for NF-κB termination. Knock-in mice that express a version of A20 lacking deubiquitylase activity have no signs of inflammation, possess a normal complement of B, T, dendritic, and myeloid cells, and undergo normal dynamics of TNF- and LPS-induced NF-κB activation.39
Figure 2.

TNFR1 signaling. Upon stimulation with TNF, TNFR1 recruits TRADD and RIPK1, followed by cIAP1, cIAP2, TRAF2, and TRAF5. RIPK1 is then K63-polyubiquitylated by cIAP1 and cIAP2, which allows docking of the TAK1–TAB2 or TAB3 complex and the IKK complex. Assembly of the IKK complex activates the NF-κB pathway, which is enhanced by recruitment of the LUBAC through RIPK1 linear ubiquitylation. Subsequently, upon internalization of TNFR1, RIPK1 is believed to dissociate from the receptor as a result of deubiquitylation by CYLD and interact with FADD, procaspase-8, and FLIPs. The long isoform of FLIP (FLIPL) and procaspase-8 form a heterodimeric caspase that cleaves and inactivates RIPK1, RIPK3, and CYLD to prevent necroptosis. This TRADD-dependent complex (also referred to as complex IIa) allows caspase-8 homodimerization and activation, which activates the executioner caspases 3 and 7 resulting in apoptosis. However, when caspase-8 is inhibited by chemical caspase inhibitors or virally encoded proteins such as CrmA or vIRA, the RHIM domains of RIPK1 and RIPK3 associate in microfilament-like complexes called necrosomes. The mutual auto- and transphosphorylation of RIPK1/3 induces the oligomerization of MLKL, which initiates necroptosis. Upon cIAP inhibition (e.g., by SMAC mimetics) a large TRADD-independent cytosolic complex is formed between RIPK1, RIPK3, FADD, and the FLIPL/caspase-8 heterodimer, which is referred to as complex IIb. Similar to complex IIa, RIPK1 and RIPK3 are also inactivated through cleavage by caspase-8/FLIPL heterodimers, apoptosis is induced by release of caspase-8 homodimers, and necroptosis is induced upon defective caspase-8 function or recruitment. Abbreviations: cIAP, cellular inhibitor of apoptosis; FADD, Fas-associated death domain; FLIP, FLICE-like inhibitory protein; IKK, inhibitor κB kinase; LUBAC, linear ubiquitin chain assembly complex; RIPK, receptor interacting protein kinase; RHIM, RIP homotypic interacting motif; MLKL, mixed lineage kinase domain-like; NF-κB, nuclear factor kappa B; SMAC, second mitochondria-derived activator of caspase; TAB, TAK1 binding protein; TAK, transforming growth factor-β–activated kinase; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TRADD, TNF receptor–associated death domain; TRAF, TNF receptor–associated factor; vIRA, viral M45-encoded inhibitor of RIP activation.
TNF-induced NF-κB activation drives the expression not only of inflammatory mediators but also of proteins directly involved in apoptosis inhibition, such as several Bcl2 family members, FLICE-like inhibitory proteins (cFLIPs), and cIAPs (Supplementary Table 1). In accordance with this, mouse embryonic fibroblasts (MEFs) derived from mice deficient in components of the NF-κB pathway, such as NEMO, IKKα/β, p65, or p50/65, fail to upregulate expression of these antiapoptotic genes and consequently undergo caspase-8–dependent apoptosis following TNF stimulation. Mice deficient in NEMO, IKKα/β, p65, or p50/65 die at embryonic stage E12-16 as a result of TNFR1-dependent apoptosis in the liver.40
RIPK1 is a crucial adapter that plays multiple roles within TNFR1-signaling complexes. RIPK1 ubiquitylation regulates both TNF-induced NF-κB activation and cell death. The ubiquitin chains conjugated to RIPK1 contribute to recruitment of the IKKα/β/NEMO complex to the TNFR1 complex I.41 TNF-induced NF-κB activation therefore fails to proceed efficiently in cells lacking RIPK1.42-44 The absence of fully defective NF-κB induction in certain cell types45 probably originates from activation of alternative mechanisms that compensate for RIPK1 deficiency.46 This “non-essential” role of RIPK1 in TNF-induced NF-κB activation is supported genetically, as RIPK1-null embryos survive until birth and therefore do not phenocopy the early embryonic lethality of mice deficient in the “essential” components of the NF-κB pathway, such as p65. Nevertheless, absence of RIPK1 still sensitizes cells to caspase-8–dependent apoptosis in response to TNF.42,44,47 These findings were recently underscored in an in vivo context in which the perinatal death of RIPK1-deficient mice is triggered, in part, by aberrant caspase-8 activation.43,48,49 RIPK1 protects cells from caspase-8–induced apoptosis by contributing to robust antiapoptotic gene induction as well as by stabilizing cFLIP levels in cells.43 The idea that the antiapoptotic function of RIPK1 is not limited to NF-κB activation is supported by an earlier in vitro study that demonstrated sensitivity of RIPK1-deficient cells to TNF-mediated death even in NF-κB inhibited conditions.50
Paradoxically, in addition its antiapoptotic function, RIPK1 also positively regulates TNF-mediated apoptosis under certain conditions. In order to understand the induction of apoptosis by TNF, it is important to discriminate between 2 major conditions: TNF signaling in the presence of protein translation inhibition by cycloheximide (CHX) and that in conditions of cIAP1/2 elimination (obtained by treatment with second mitochondria-derived activator of caspase [SMAC] mimetics or physiologically upon activation of certain TNFRs, such as CD40, BAFFR, TNFRSF12A, and LTβR). When the antiapoptotic NF-κB response is inhibited by CHX, TNFR1 ligation switches from a prosurvival to a proapoptotic response. This switch occurs via internalization of complex I and assembly of complex IIa (Fig. 2).33 Translation repression promotes caspase-8 activation by reducing the levels of cFLIP, a relatively unstable endogenous caspase-8 inhibitor. Under these conditions, RIPK1 deficiency further sensitizes the cells to death.50,51
The E3 ubiquitin ligase activities of cIAP1/2 are required for TNF-induced canonical NF-κB activation.52-54 Consequently, their depletion also induces a switch to caspase-8–mediated apoptosis. Of note, TNF-induced apoptosis in the absence of cIAP1/2 occurs more rapidly and at a higher rate than that induced upon single NF-κB inhibition, indicating that cIAP1/2 additionally regulates an NF-κB–independent cell death checkpoint in the TNFR1.51,55 Interestingly, the excessive death obtained under these conditions was shown to rely on RIPK1 kinase activity and not on TRADD (in contrast to complex IIa).51,56 The RIPK1-containing cytosolic death complex obtained in cIAP1/2-depleted conditions has been defined as complex IIb to discriminate it from complex IIa (Fig. 2).34,57 The molecular mechanism accounting for the differential assembly of complex IIa versus IIb is poorly understood, but is suggested to rely on the differential ubiquitylation status of RIPK1 in complex I. Indeed, cIAP1/2 directly conjugates RIPK1 with ubiquitin chains,52,53,58 and cIAP1/2-mediated RIPK1 ubiquitylation in complex I is believed to prevent RIPK1 from integrating complex II.52,56,59 This notion is supported by the fact that repression of the RIPK1 deubiquitylase CYLD inhibits recruitment of RIPK1 to complex IIb.56 Nevertheless, recent studies indicate that it is probably the proteins recruited to the ubiquitin chains, rather than the ubiquitin chains themselves, that regulate the contribution of RIPK1 to the death trigger. Indeed, both TAK1 and NEMO depletion also induce complex IIb-mediated apoptosis without affecting RIPK1 ubiquitylation.51,60 Recently, Pellino3 was also found to negatively control formation of complex IIb in a NF-κB– and ubiquitin ligase-independent manner.61
In summary, TNFR1 primarily induces formation of a membrane-bound complex that triggers NF-κB– and MAPK-mediated gene activation and negatively controls formation of the cytosolic death inducing complex II. Toggling of TNFR1-signaling to RIPK1-dependent apoptosis requires a secondary signal and prevails only upon interference with the primary signal, for example by depletion of cIAPs, cFLIPL, TAK1, NEMO, or Pellino3 (Fig. 2).
Fas-induced cell death versus gene activation
Signaling downstream of FAS and TRAILR1/2 also involves assembly of primary and secondary complexes but the respective function of these complexes is reversed compared to TNFR1 signaling (Fig. 1). Indeed, the FAS- and TRAILR1/2-associated primary complex (known as DISC) is composed of the adaptor FADD, caspase-8, and the cFLIP-isoforms, and is responsible for caspase-8–dependent apoptosis.62 Note that RIPK1 has also been shown to directly bind to FAS and mediate cell death in the absence of cIAPs.63,64 This binding seems to be favored mainly when caspases are blocked.65 A secondary cytosolic gene-activating complex originates from dissociation of this membrane-bound complex.66,67 In the case of TRAILR1/2, release of FADD from the DISC triggers formation of a cytosolic complex II that recruits TRAF2, cIAP1/2, RIPK1, NEMO, and most likely several other factors to activate NF-κB, MAPKs, and consequently gene induction.68 In line with these findings, FAS-induced apoptosis or necroptosis can also be associated with the production of cytokines and chemokines, which requires RIPK1 and cIAP1/2 for optimal production.69,70 The chemotaxis of phagocytes toward apoptotic cells suggests that these induced factors function as additional “find-me” signals. Interference with caspase activation (primary signal) through chemical-70 or pathogen-71 induced inhibition enhances gene activation (secondary signal). In addition, and contrary to TNFR1 complex I, Fas and TRAILR1/2 complex II were also reported to contribute to cell death under certain conditions by enhancing autocatalytic activation of caspase-842 or by inducing gene activation (Fig. 1).62,71
Interplay Between Apoptosis and Necroptosis Induction Downstream of TNFR1
Having outlined the mutual interplay between survival and cell death signaling downstream of TNFR1 activation, we now describe the interplay between factors contained within the cytosolic death complex that dictate the apoptotic or necroptotic outcomes.
Caspase-8/FLIP
Within the death inducing complex II, caspase-8 activity controls apoptotic and necroptotic cell fates. Active caspase-8 suppresses necroptosis by cleaving several substrates, including RIPK1, RIPK3, and CYLD.72-74 CYLD-mediated deubiquitylation of RIPK1 within complex II was reported to facilitate RIPK1 kinase activation and subsequent necroptosis induction.75 Vice versa, CYLD is downregulated in a TLR-dependent way in wild-derived mice to protect macrophages from necroptosis.76 The proapoptotic/antinecroptotic function of caspase-8 is regulated by specific interactions with the pseudo-caspase cFLIP. Whereas caspase-8 homodimers engage apoptosis, caspase-8/cFLIPL heterodimers suppress both apoptosis and necroptosis.77 Binding of cFLIPL to caspase-8 throttles caspase-8 activity to prevent apoptosis but allows low levels of enzymatic activity sufficient to cleave and inactivate RIPK1 and RIPK3 (Fig. 3).78 In cells expressing adequate levels of the pronecrotic kinase RIPK3, TNF-signaling triggers necroptosis after elimination of this basal caspase-8 activity.79 In accordance with these findings, the embryonic lethality of mice deficient in caspase-8 or FADD can be completely rescued by ablation of RIPK3 or RIPK1 (Supplementary Table 1).80-83 It remains unclear what regulates the different outcomes of caspase-8 homodimer versus caspase-8/cFLIPL heterodimer activity, but possible mechanisms include differences in cleavage specificities, access to specific substrates, level of activity, or subcellular localization.84 Interestingly, caspase-8 heterodimerization with an alternative form of cFLIP, cFLIPS, prevents caspase-8 activity but enhances complex II formation to favor necroptosis induction (Fig. 3).65,85 Together, these results indicate that the amount of cFLIP in cells protects them from extrinsic apoptosis and that the ratio of cFLIPL and cFLIPS isoforms determines the sensitivity to necroptosis. This spectrum of cell death regulatory mechanisms is not restricted to TNF signaling but also operates to control cell fate following DNA damage or TLR signaling.85,86
Figure 3.
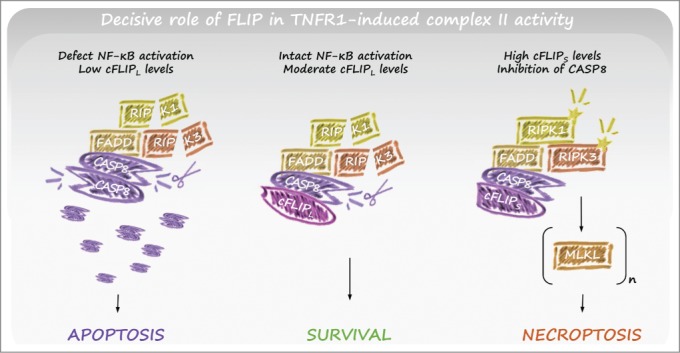
Role of FLIP in TNFR1-induced complex II activity. TNFR1 stimulation typically induces NF-κB–mediated survival signaling, for example through upregulation of FLIPL (central panel). Caspase-8/FLIPL heterodimers allow local caspase-8 activity within complex II, resulting in cleavage of RIPK1 and RIPK3. As a result, apoptosis and necroptosis are inhibited. However, when FLIPL levels are low (for example due to defective NF-kB activation) active caspase-8 homodimers form, are released from complex II, and induce apoptosis (left panel). In conditions where FLIPs levels are upregulated, caspase-8/FLIPs heterodimers inhibit local caspase-8 activity, allowing RIPK1/3-mediated necroptosis (right panel). Abbreviations: FLIP, FLICE-like inhibitory protein; NF-κB, nuclear factor kappa B; RIPK, receptor interacting protein kinase; TNFR, tumor necrosis factor receptor.
RIPK1
The complex role that RIPK1 plays in regulating caspase-8 and RIPK3 was in part revealed by the recent demonstration that the perinatal lethality of RIPK1-null mice was due to a combination of aberrant activation of caspase-8 and RIPK3, such that mice lacking all 3 enzymes survived to adulthood.43,48,49 Both cell autonomous and non-cell autonomous effects contribute to the necessity for RIPK1 for mammalian life. On one hand RIPK1 is required to prevent apoptosis induced by TNF, possibly related to a failure to stabilize and upregulate cFLIPL in response to TNFR1 signaling. This is recently underscored by findings that mice lacking RIPK1 specifically in the intestinal epithelium developed severe inflammatory bowel disease within the first weeks of life due to extensive caspase-8 mediated apoptosis, and died as a result.87,88 On the other hand, RIPK1 prevents RIPK3-driven necroptosis promoted by interferon (IFN) and the TLR-adapter TRIF,43 and possibly other signals. Since RIPK1 is reported to be essential for RIPK3 activation and subsequent necroptosis induction by TNF, it is surprising to identify settings where RIPK1 actively suppresses RIPK3. Whereas RIPK1 is upstream of RIPK3 for TNF-induced necroptosis, RIPK3 is apical to RIPK1 in TLR- and IFN-induced necroptosis. In the context where the ordering of kinases is reversed, RIPK1 may function as an adapter to bridge RIPK3 to the necroptosis suppressive activity of caspase-8, and in the absence of RIPK1 developmental cues might drive lethal levels of RIPK3 activity. The dynamic interplay and interdependence of these complex II components confers a crucial host defense function to limit pathogen spread, especially upon interference or perturbation of any one of these processes.89,90 This might explain why this complex interrelationship exists and why ablation of specific elements (such as RIPK1, FADD, caspase-8, and cFLIP) pushes the system to lethality. In line with this reasoning, the tissues most affected by disruption of these gene products (intestine, lung, skin, endothelium, hematopoietic cells) represent crucial barriers to infection that are constantly engaged by pathogens.91
The central role of RIPK1 kinase activity in deleterious TNF signaling was recently revealed genetically by the generation of RIPK1 kinase dead knock-in mice.49,92-94 In contrast to RIPK1-null mice, which die within a few days after birth, RIPK1 kinase dead knock-in mice are viable and fertile, demonstrating that the vital role of RIPK1 following mammalian parturition is due to RIPK1 adapter function rather than enzymatic activity. Nevertheless, mice expressing a kinase dead mutant of RIPK1 were completely resistant to TNF-induced shock and, as anticipated, cells derived from these mice were protected from TNF-induced cell death (Fig. 4).49,92-94 In addition, RIPK1 kinase activity was dispensable for MAPK and NF-κB activation, indicating that apoptosis/necroptosis mediated by RIPK1 enzymatic activity is probably the dominant driver of TNF-shock rather than primary gene induction.
Figure 4.
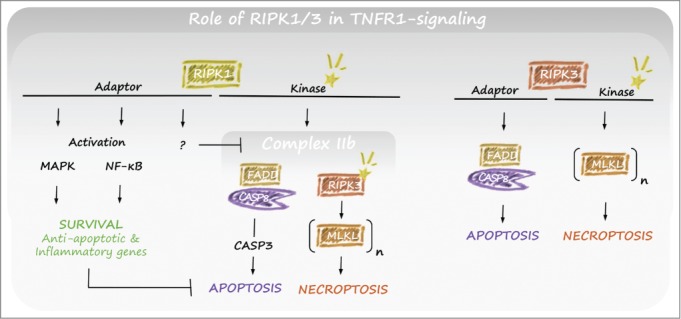
Role of RIPK1/3 in TNFR1-induced complex II activity. RIPK1 signals toward survival and cell death through different functional domains. The adaptor function of RIPK1 is important for activation of MAPK and NF-κB resulting in cell death inhibition, and has a NF-κB–independent inhibitory action on the formation of complex IIb. The kinase activity of RIPK1 can be involved in both necroptosis and apoptosis within complex II. Typically, this occurs under conditions when cIAP1/2 or TAK1 is degraded. RIPK3 is primarily involved in the induction of cell death. Although its kinase is typically involved in the induction of necroptosis, its adaptor function can also contribute to the induction of apoptosis. Abbreviations: cIAP, cellular inhibitor of apoptosis; FLIP, FLICE-like inhibitory protein; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; RIPK, receptor interacting protein kinase; TAK, transforming growth factor-β–activated kinase; TNFR, tumor necrosis factor receptor.
Mice lacking Sharpin, a component of the LUBAC complex, develop a lethal inflammatory skin phenotype within 12 weeks of age and also manifest multiorgan inflammation. This severe TNF-mediated inflammatory phenotype is absent in Sharpin mutant mice bred on a RIPK1 kinase dead background, demonstrating the central role of aberrant RIPK1 kinase activity in this animal model.92 Together, these findings clearly demonstrate a tissue-specific requirement for RIPK1 kinase activity upstream of both caspase-8 and RIPK3 in promoting inflammation. Of note, patients with mutations in HOIL, another LUBAC component, also develop severe autoinflammation that is probably driven by RIPK1 kinase activity.95 It will be important to evaluate whether a deficiency in RIPK1 kinase activity also rescues the lethal phenotype of mice deficient in cIAP1/2, FADD, caspase-8, or cFLIP, which succumb at E10.5 due to TNF-induced necroptosis and apoptosis, and of mice deficient in p65, IκB, IKKβ, NEMO, or TAB2, which die later in gestation due to TNF-induced liver apoptosis. Thus, as a key regulator of inflammation, apoptosis, and necroptosis, RIPK1 is positioned at a strategic crossroads of multiple signaling nodes in the inflammatory response and must be tightly regulated to ensure normal tissue homeostasis.
RIPK3
Although RIPK3 knockout mice are viable and fertile, RIPK3 D161N kinase dead knock-in mice die at embryonic day E10.5 due to massive levels of apoptosis in the embryo and yolk sac vasculature. This embryonic death is rescued by ablation of RIPK1 or caspase-8, indicating that RIPK3 can itself engage both RIPK1 and caspase-893 and suggesting that RIPK3 kinase activity is essential in embryos to prevent aberrant complex II formation and lethal apoptosis. It remains unclear at the structural level why D161N kinase-dead RIPK3 is proapoptotic, though it is likely that the kinase domain functionally “masks” the RIP homotypic interacting motif (RHIM) to prevent spurious activation.96 In this scenario, D161N alters the conformation of RIPK3 such that the RHIM is exposed for binding to RIPK1 to initiate apoptosis. This model predicts that the kinase and RHIM domains collaborate to control scaffolding of the necroptotic and apoptotic machineries. Whereas TNF signaling typically requires RIPK1 to activate RIPK3 to induce necroptosis, it has been noted that TNF can trigger RIPK3 activation even in the absence of RIPK1 if RIPK3 levels are induced to high levels in cells (Fig. 4).97 In the absence of RIPK1 and the presence of elevated levels of RIPK3, TNF can activate RIPK3 to induce cell death by both a caspase-8–dependent mechanism and a caspase-independent mechanism.51,97 Moreover, RIPK3 has also been reported to positively contribute to full caspase-8 activation downstream of TNFR1 independently of its kinase activity or intact RHIM in conditions of RIPK1-dependent apoptosis.51 Finally, similar to depletion of RIPK1,44,47 blocking TNF-induced necroptosis by suppressing RIPK3 or MLKL toggles the cell death response to apoptosis, albeit with different kinetics.98 Collectively, these studies indicate that the precise control of the complex II machinery is necessary to prevent a lethal imbalance of necroptotic or apoptotic pathways.
Emerging Insights in Pathogen-Cell Death Interactions
Considering the crucial role of DRs in homeostasis and their therapeutic impact (for example, anti-TNF drugs such as Enbrel®, Remicade®, Humira®, Cimzia®, and Simponi®), most of our molecular understanding of cell fate originates from DR-focused research. However, new exciting fields that are interconnected with cell death beyond DRs are now being explored. In this respect, we are beginning to pinpoint bottlenecks or converging nodes in cell death signaling that can be therapeutically explored. In particular, much insight has been gained through the study of pathogens, such as herpes virus, that have evolved mechanisms to evade cell death defenses over 100 million years. A prototype example is cytomegalovirus (CMV), which suppresses caspase-8-induced apoptosis and RIPK3-induced necroptosis through genes encoding vICA and vIRA respectively (reviewed extensively elsewhere71,90). These findings provide key insights into how inhibition of caspase-8 by a viral gene unleashes RIPK3-mediated necroptosis that is in turn suppressed by a counter adaptation to close the antiviral necroptotic trap door (see Mocarski et al. for more extended reading99).
Concluding Perspectives
Intense research on inflammation and cell death indicates that regulated necrosis is an important cause or catalyst of several common diseases,4 putting cell death research once more in the center stage. Current therapeutic strategies mainly focus on inhibition of the kinase activity of RIPK1 and RIPK3. Until recently, RIPK3 inhibitors were predicted to be superior over RIPK1 inhibitors because necroptosis occurs in the absence of RIPK1, but not in the absence of RIPK3, in response to some stimuli.79,97,100 However, the lethal phenotype of the RIPK3 D161N kinase dead knock-in mice combined with the capacity of small-molecule inhibitors of RIPK3 to spontaneously induce apoptosis suggest that pharmacologic targeting of RIPK3 will remain a challenge.18 RIPK1 kinase-deficient mice, however, reveal no spontaneous phenotype and retain the ability to activate NF-κB and MAPK pathways despite a defect in both apoptotic and necroptotic cell death.49,92-94 These findings have re-energized interest in targeting the kinase function of RIPK1. Additionally, RIPK1 kinase inhibition may preserve beneficial inflammation while preventing the deleterious outcomes of inflammation, which might enhance its therapeutic impact. Alternatively, small molecules that disrupt RHIM signaling could also be effective at preventing cell death but so far such molecules have remained elusive.
A reviving field, originally recognized by Virchow, is the close interconnection between cell death and inflammation. This is emphasized by some recent findings that classic cell death inducers such as caspase-8 and RIPK3 seem to act upstream of inflammasome activation in a cell autonomous way.101-106 However, the mechanisms of this connection remain unclear. RIPK1, RIPK3, and other complex II components have all been implicated in the regulation of inflammation although uncoupling these functions from their roles in cell death remains a challenge. This could imply that inducers of the classic prototypic cell death types of apoptosis and necroptosis are involved in the switch to more professional cell death mechanisms, such as pyroptosis and maybe also (N)ETosis, as a defense strategy against pathogens. Clarifying the latter issue will be very important in the light of current therapeutic strategies to explore the potential of blocking necrosome functioning in an attempt to interfere with degenerative diseases.
Despite recent advances in our understanding of inflammation, apoptosis, necroptosis, and their entwined relationships, many unanswered questions still need to be addressed. An intriguing aspect that requires further clarification is the NF-κB–independent inhibitory action of complex I members in repressing complex II activation. RIPK1, TAK1, NEMO, and TAB2 were proposed as candidates that directly control complex II formation or activity. In particular, the fact that TAB2-deficient mice do not show a phenotype on NF-κB activation107 yet die from massive liver apoptosis similar to mice deficient in p65, IKKβ, TAK1, or NEMO mice remains intriguing and incompletely understood. Moreover, the rescue of RIPK1 kinase dead mutant mice from both TNF-induced shock and the lethal TNF-induced inflammation in Sharpin mutant mice call into question the dominance of NF-κB and MAPK activation in deleterious inflammation and suggest that RIPK-kinase dependent processes including both apoptosis and necroptosis may be the major TNF-signaling outputs contributing to diseases caused by this master regulator.
What exactly triggers the conversion of complex I to complex II? Is it internalization, deubiquitylase activity, or still unidentified mechanisms? The proposed role of CYLD in the transition of complex I to II56 has recently been questioned; instead, CYLD function was proposed to act mainly within complex II itself.75 There is accumulating evidence that internalization is required to allow the induction of cell death,108-110 which would imply subsequent formation of complex II. The recent findings that mice deficient in ceramide synthase 2 are completely protected against lipopolysaccharide/Gal-induced hepatic liver failure due to defect TNFR internalization108 and that viruses interfere with DR internalization to block cell death induction109 re-enforce the central role of internalization in the induction of cell death. It would be interesting to analyze the formation of complex II in cells deficient in TNFR internalization to clarify this issue. Alternatively, considering that depletion of IAPs by SMAC mimetics triggers complex II formation in some cells independent of TNF-signaling, and thus complex I, the cytosolic complex II may form sequentially and not as a direct maturation of complex I.
In conclusion, the mutual negative interplay between cell death pathways nicely illustrates the integration of biological processes. One trigger initiates a spectrum of competing signals. This signaling cross talk provides the capacity to survey transduction and provide alternative outcomes to contend with situations such as interference by pathogens. Unraveling these highly interconnected host defense networks will remain a challenge but should provide a path forward for the treatment of inflammatory disease. Although this review has focused primarily on the relationship between apoptosis and necroptosis, further efforts will likely unveil unexpected connections to other cell death processes such as autophagy, organelle stress, lipid metabolism, and senescence, and provide insights into the design of therapeutic strategies that can target “bad” inflammation while preserving “good” inflammation.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors apologize to colleagues whose work was not discussed owing to space limitations.
Funding
TV is supported by a research grant from the Foundation against Cancer (2012-188). MB has a tenure track position within the Multidisciplinary Research Program of Ghent University (MRP, GROUP-ID consortium). Research in the Vandenabeele group is further supported by European grants (FP6 ApopTrain, MRTNCT-035624; FP7 EC RTD Integrated Project, Apo-Sys, FP7-200767; Euregional PACT II), Belgian grants (Interuniversity Attraction Poles, IAP 6/18, IAP 7/32), Flemish grants (Research Foundation Flanders, FWO G.0875.11, FWO G.0973.11 N, FWO G.0A45.12 N, FWO G.0172.12, FWO G.0787.13N, G0C3114N and FWO KAN 31528711), Ghent University grants (MRP, GROUP-ID consortium), and grants from Flanders Institute for Biotechnology (VIB). PV holds a Methusalem grant (BOF09/01M00709) from the Flemish Government.
References
- 1.Virchow R. Cellular Pathology: As Based Upon Physiological and Pathological Histology. Twenty Lectures Delivered in the Pathological Institute of Berlin During the Months of February, March and April, 1858, (RM De Witt, 1860). [Google Scholar]
- 2.Wallach D, Kang TB, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol 2014; 14:51–9; PMID:24336099; http://dx.doi.org/ 10.1038/nri3561 [DOI] [PubMed] [Google Scholar]
- 3.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014; 15:135–47; PMID:24452471; http://dx.doi.org/ 10.1038/nrm3737 [DOI] [PubMed] [Google Scholar]
- 4.Linkermann A, Vanden Berghe T, Takahashi N, Kunzendorf U, Krautwald S, Vandenabeele P. The Potential Role of Necroptosis in Diseases In Necrotic Cell Death. eds. Shen HM, Vandenabeele P. 1–22, New York: Humana Press, Springer; 2014. [Google Scholar]
- 5.Lockshin RA, Williams CM. Programmed cell death. II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 1964; 10:643–9; http://dx.doi.org/ 10.1016/0022-1910(64)90034-4 [DOI] [Google Scholar]
- 6.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26:239–57; PMID:4561027; http://dx.doi.org/ 10.1038/bjc.1972.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology 1973; 7:253–66; PMID:4807128; http://dx.doi.org/ 10.1002/tera.1420070306 [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al.. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107–20; PMID:21760595; http://dx.doi.org/ 10.1038/cdd.2011.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol 2006; 7:97–108; PMID:16493416; http://dx.doi.org/ 10.1038/nrm1836 [DOI] [PubMed] [Google Scholar]
- 10.Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 1988; 141:2629–34; PMID:3171180 [PubMed] [Google Scholar]
- 11.Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 2006; 1757:1371–87; PMID:16950166; http://dx.doi.org/ 10.1016/j.bbabio.2006.06.014 [DOI] [PubMed] [Google Scholar]
- 12.Van Loo G, Demol H, van Gurp M, Hoorelbeke B, Schotte P, Beyaert R, Zhivotovsky B, Gevaert K, Declercq W, Vandekerckhove J, et al.. A matrix-assisted laser desorption ionization post-source decay (MALDI-PSD) analysis of proteins released from isolated liver mitochondria treated with recombinant truncated Bid. Cell Death Differ 2002; 9:301–8; PMID:11859412; http://dx.doi.org/ 10.1038/sj.cdd.4400966 [DOI] [PubMed] [Google Scholar]
- 13.van Loo G, van Gurp M, Depuydt B, Srinivasula SM, Rodriguez I, Alnemri ES, Gevaert K, Vandekerckhove J, Declercq W, Vandenabeele P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ 2002; 9:20–6; PMID:11803371; http://dx.doi.org/ 10.1038/sj.cdd.4400970 [DOI] [PubMed] [Google Scholar]
- 14.Linkermann A, Green DR. Necroptosis. N Engl J Med 2014; 370:455–65; PMID:24476434; http://dx.doi.org/ 10.1056/NEJMra1310050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou W, Yuan J. SnapShot: Necroptosis. Cell 2014; 158:464–40; PMID:25036639; http://dx.doi.org/ 10.1016/j.cell.2014.06.041 [DOI] [PubMed] [Google Scholar]
- 16.Zhou W, Yuan J. Necroptosis in health and diseases. Semin Cell Dev Biol 2014; 35:14–23; PMID:25087983 [DOI] [PubMed] [Google Scholar]
- 17.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1:112–9; PMID:16408008; http://dx.doi.org/ 10.1038/nchembio711 [DOI] [PubMed] [Google Scholar]
- 18.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 2013; 288:31268–79; PMID:24019532; http://dx.doi.org/ 10.1074/jbc.M113.462341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al.. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148:213–27; PMID:22265413; http://dx.doi.org/ 10.1016/j.cell.2011.11.031 [DOI] [PubMed] [Google Scholar]
- 20.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al.. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 2014; 7:971–81; PMID:24813885; http://dx.doi.org/ 10.1016/j.celrep.2014.04.026 [DOI] [PubMed] [Google Scholar]
- 21.Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. A Plug Release Mechanism for Membrane Permeation by MLKL. Structure 2014; 22:1489–500; PMID:25220470; http://dx.doi.org/ 10.1016/j.str.2014.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54:133–46; PMID:24703947; http://dx.doi.org/ 10.1016/j.molcel.2014.03.003 [DOI] [PubMed] [Google Scholar]
- 23.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al.. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156:317–31; PMID:24439385; http://dx.doi.org/ 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al.. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149:1060–72; PMID:22632970; http://dx.doi.org/ 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012; 13:411–24; PMID:22713970; http://dx.doi.org/ 10.1038/nrm3376 [DOI] [PubMed] [Google Scholar]
- 26.Croker BA, O'Donnell JA, Gerlic M. Pyroptotic death storms and cytopenia. Curr Opin Immunol 2014; 26:128–37; PMID:24556409; http://dx.doi.org/ 10.1016/j.coi.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 27.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Muñoz-Arias I, et al.. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014; 505:509–14; PMID:24356306; http://dx.doi.org/ 10.1038/nature12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014; 343:428–32; PMID:24356113; http://dx.doi.org/ 10.1126/science.1243640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldmann O, Medina E. The expanding world of extracellular traps: not only neutrophils but much more. Frontiers in immunology 2012; 3:420; PMID:23335924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl) 2014; 92:465–72; PMID:24706102; http://dx.doi.org/ 10.1007/s00109-014-1148-z [DOI] [PubMed] [Google Scholar]
- 31.Yipp BG, Kubes P. NETosis: how vital is it? Blood 2013; 122:2784–94; PMID:24009232; http://dx.doi.org/ 10.1182/blood-2013-04-457671 [DOI] [PubMed] [Google Scholar]
- 32.Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol 2013; 5:a008698; PMID:23637280; http://dx.doi.org/ 10.1101/cshperspect.a008698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114:181–90; PMID:12887920; http://dx.doi.org/ 10.1016/S0092-8674(03)00521-X [DOI] [PubMed] [Google Scholar]
- 34.Estornes Y, Bertrand MJ. IAPs, regulators of innate immunity and inflammation. Semin Cell Dev Biol 2014; S1084-9521:00073–1; PMID:24718315; http://dx.doi.org/ 10.1016/j.semcdb.2014.03.035 [DOI] [PubMed] [Google Scholar]
- 35.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 2011; 12:715–23; PMID:21772280; http://dx.doi.org/ 10.1038/ni.2060 [DOI] [PubMed] [Google Scholar]
- 36.Xu G, Tan X, Wang H, Sun W, Shi Y, Burlingame S, Gu X, Cao G, Zhang T, Qin J, et al.. Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor alpha-induced nuclear factor kappaB activation via binding to and deubiquitinating receptor-interacting protein 1. J Biol Chem 2010; 285:969–78; PMID:19910467; http://dx.doi.org/ 10.1074/jbc.M109.042689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al.. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004; 430:694–9; PMID:15258597; http://dx.doi.org/ 10.1038/nature02794 [DOI] [PubMed] [Google Scholar]
- 38.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000; 289:2350–4; PMID:11009421; http://dx.doi.org/ 10.1126/science.289.5488.2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De A, Dainichi T, Rathinam CV, Ghosh S. The deubiquitinase activity of A20 is dispensable for NF-kappaB signaling. EMBO Rep 2014; 15:775–83; PMID:24878851; http://dx.doi.org/ 10.15252/embr.201338305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou A, Scoggin S, Gaynor RB, Williams NS. Identification of NF-kappa B-regulated genes induced by TNFalpha utilizing expression profiling and RNA interference. Oncogene 2003; 22:2054–64; PMID:12673210; http://dx.doi.org/ 10.1038/sj.onc.1206262 [DOI] [PubMed] [Google Scholar]
- 41.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22:245–57; PMID:16603398; http://dx.doi.org/ 10.1016/j.molcel.2006.03.026 [DOI] [PubMed] [Google Scholar]
- 42.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998; 8:297–303; PMID:9529147; http://dx.doi.org/ 10.1016/S1074-7613(00)80535-X [DOI] [PubMed] [Google Scholar]
- 43.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, et al.. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014; 157:1189–202; PMID:24813850; http://dx.doi.org/ 10.1016/j.cell.2014.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2011; 2:e230; PMID:22089168; http://dx.doi.org/ 10.1038/cddis.2011.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong WW, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ 2010; 17:482–7; PMID:19927158; http://dx.doi.org/ 10.1038/cdd.2009.178 [DOI] [PubMed] [Google Scholar]
- 46.Blackwell K, Zhang L, Workman LM, Ting AT, Iwai K, Habelhah H. Two coordinated mechanisms underlie tumor necrosis factor alpha-induced immediate and delayed IκB kinase activation. Mol Cell Biol 2013; 33:1901–15; PMID:23459942; http://dx.doi.org/ 10.1128/MCB.01416-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P. Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem 2003; 278:5622–9; PMID:12441346; http://dx.doi.org/ 10.1074/jbc.M208925200 [DOI] [PubMed] [Google Scholar]
- 48.Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, et al.. RIPK1 regulates RIPK3-MLKL-Driven systemic inflammation and emergency hematopoiesis. Cell 2014; 157:1175–88; PMID:24813849; http://dx.doi.org/ 10.1016/j.cell.2014.04.019 [DOI] [PubMed] [Google Scholar]
- 49.Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, et al.. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A 2014; 111:7753–8; PMID:24821786; http://dx.doi.org/ 10.1073/pnas.1401857111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gentle IE, Wong WW, Evans JM, Bankovacki A, Cook WD, Khan NR, Nachbur U, Rickard J, Anderton H, Moulin M, et al.. In TNF-stimulated cells, RIPK1 promotes cell survival by stabilizing TRAF2 and cIAP1, which limits induction of non-canonical NF-kappaB and activation of caspase-8. J Biol Chem 2011; 286:13282–91; PMID:21339290; http://dx.doi.org/ 10.1074/jbc.M110.216226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJ. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 2013; 20:1381–92; PMID:23892367; http://dx.doi.org/ 10.1038/cdd.2013.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30:689–700; PMID:18570872; http://dx.doi.org/ 10.1016/j.molcel.2008.05.014 [DOI] [PubMed] [Google Scholar]
- 53.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem 2008; 283:24295–9; PMID:18621737; http://dx.doi.org/ 10.1074/jbc.C800128200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, et al.. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A 2008; 105:11778–83; PMID:18697935; http://dx.doi.org/ 10.1073/pnas.0711122105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O'Donnell MA, Ting AT. NFκB and ubiquitination: partners in disarming RIPK1-mediated cell death. Immunol Res 2012; 54:214–26; PMID:22477525; http://dx.doi.org/ 10.1007/s12026-012-8321-7 [DOI] [PubMed] [Google Scholar]
- 56.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133:693–703; PMID:18485876; http://dx.doi.org/ 10.1016/j.cell.2008.03.036 [DOI] [PubMed] [Google Scholar]
- 57.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol 2009; 10:348–55; PMID:19295631; http://dx.doi.org/ 10.1038/ni.1714 [DOI] [PubMed] [Google Scholar]
- 58.Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael-Tomasevic A, Phu L, Helgason E, Fairbrother WJ, Deshayes K, Kirkpatrick DS, et al.. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J 2010; 29:4198–209; PMID:21113135; http://dx.doi.org/ 10.1038/emboj.2010.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Donnell MA, Legarda-Addison D, Skountzos P, Yeh WC, Ting AT. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol 2007; 17:418–24; PMID:17306544; http://dx.doi.org/ 10.1016/j.cub.2007.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Legarda-Addison D, Hase H, O'Donnell MA, Ting AT. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ 2009; 16:1279–88; PMID:19373245; http://dx.doi.org/ 10.1038/cdd.2009.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang S, Wang B, Tang LS, Siednienko J, Callanan JJ, Moynagh PN. Pellino3 targets RIP1 and regulates the pro-apoptotic effects of TNF-alpha. Nat Commun 2013; 4:2583; PMID:24113711; http://dx.doi.org/ 10.1038/ncomms3583 [DOI] [PubMed] [Google Scholar]
- 62.Sessler T, Healy S, Samali A, Szegezdi E. Structural determinants of DISC function: new insights into death receptor-mediated apoptosis signalling. Pharmacol Ther 2013; 140:186–99; PMID:23845861; http://dx.doi.org/ 10.1016/j.pharmthera.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 63.Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 1995; 81:513–23; PMID:7538908; http://dx.doi.org/ 10.1016/0092-8674(95)90072-1 [DOI] [PubMed] [Google Scholar]
- 64.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1:489–95; PMID:11101870; http://dx.doi.org/ 10.1038/82732 [DOI] [PubMed] [Google Scholar]
- 65.Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B, Gollnick H, Silke J, Leverkus M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol 2009; 187:1037–54; PMID:20038679; http://dx.doi.org/ 10.1083/jcb.200904158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lavrik IN, Mock T, Golks A, Hoffmann JC, Baumann S, Krammer PH. CD95 stimulation results in the formation of a novel death effector domain protein-containing complex. J Biol Chem 2008; 283:26401–8; PMID:18635548; http://dx.doi.org/ 10.1074/jbc.M800823200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin Z, El-Deiry WS. Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol Cell Biol 2006; 26:8136–48; PMID:16940186; http://dx.doi.org/ 10.1128/MCB.00257-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D, Ashkenazi A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem 2005; 280:40599–608; PMID:16227629; http://dx.doi.org/ 10.1074/jbc.M509560200 [DOI] [PubMed] [Google Scholar]
- 69.Cullen SP, Henry CM, Kearney CJ, Logue SE, Feoktistova M, Tynan GA, Lavelle EC, Leverkus M, Martin SJ. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol Cell 2013; 49:1034–48; PMID:23434371; http://dx.doi.org/ 10.1016/j.molcel.2013.01.025 [DOI] [PubMed] [Google Scholar]
- 70.Vanden Berghe T, Kalai M, Denecker G, Meeus A, Saelens X, Vandenabeele P. Necrosis is associated with IL-6 production but apoptosis is not. Cell Signal 2006; 18:328–35; PMID:16023831; http://dx.doi.org/ 10.1016/j.cellsig.2005.05.003 [DOI] [PubMed] [Google Scholar]
- 71.Giogha C, Lung TW, Pearson JS, Hartland EL. Inhibition of death receptor signaling by bacterial gut pathogens. Cytokine Growth Factor Rev 2014; 25:235–43; PMID:24440054; http://dx.doi.org/ 10.1016/j.cytogfr.2013.12.012 [DOI] [PubMed] [Google Scholar]
- 72.Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 2007; 19:2056–67; PMID:17644308; http://dx.doi.org/ 10.1016/j.cellsig.2007.05.016 [DOI] [PubMed] [Google Scholar]
- 73.O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol 2011. 13:1437–42; PMID:22037414; http://dx.doi.org/ 10.1038/ncb2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rebe C, Cathelin S, Launay S, Filomenko R, Prévotat L, L'Ollivier C, Gyan E, Micheau O, Grant S, Dubart-Kupperschmitt A, et al.. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood 2007; 109:1442–50; PMID:17047155; http://dx.doi.org/ 10.1182/blood-2006-03-011585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 2013; 8:e76841; PMID:24098568; http://dx.doi.org/ 10.1371/journal.pone.0076841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schworer SA, Smirnova II, Kurbatova I, Bagina U, Churova M, Fowler T, Roy AL, Degterev A, Poltorak A. Toll-like receptor-mediated downregulation of the deubiquitinase CYLD protects macrophages from necroptosis in wild-derived mice. J Biol Chem 2014; 289:14422–33; PMID:24706750; http://dx.doi.org/ 10.1074/jbc.M114.547547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Piao X, Komazawa-Sakon S, Nishina T, Koike M, Piao JH, Ehlken H, Kurihara H, Hara M, Van Rooijen N, Schütz G, et al.. c-FLIP maintains tissue homeostasis by preventing apoptosis and programmed necrosis. Sci Signal 2012; 5:ra93; PMID:23250397; http://dx.doi.org/ 10.1126/scisignal.2003558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Salvesen GS, Walsh CM. Functions of caspase 8: The identified and the mysterious. Semin Immunol 2014; 26:246–52; PMID:24856110; http://dx.doi.org/ 10.1016/j.smim.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137:1100–11; PMID:19524512; http://dx.doi.org/ 10.1016/j.cell.2009.05.021 [DOI] [PubMed] [Google Scholar]
- 80.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep 2012; 1:401–7; PMID:22675671; http://dx.doi.org/ 10.1016/j.celrep.2012.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011; 471:368–72; PMID:21368762; http://dx.doi.org/ 10.1038/nature09857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471:363–7; PMID:21368763; http://dx.doi.org/ 10.1038/nature09852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011; 471:373–6; PMID:21368761; http://dx.doi.org/ 10.1038/nature09878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oberst A, Green DR. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol 2011; 12:757–63; PMID:22016059; http://dx.doi.org/ 10.1038/nrm3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43:449–63; PMID:21737330; http://dx.doi.org/ 10.1016/j.molcel.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, et al.. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43:432–48; PMID:21737329; http://dx.doi.org/ 10.1016/j.molcel.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 87.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, et al.. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014; 513:90–4; PMID:25132550; http://dx.doi.org/ 10.1038/nature13608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, Gonçalves A, Sze M, Gilbert B, Kourula S, et al.. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 2014; 513:95–9; PMID:25186904; http://dx.doi.org/ 10.1038/nature13706 [DOI] [PubMed] [Google Scholar]
- 89.Upton JW, Chan FK. Staying alive: cell death in antiviral immunity. Mol Cell 2014; 54:273–80; PMID:24766891; http://dx.doi.org/ 10.1016/j.molcel.2014.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaiser WJ, Upton JW, Mocarski ES. Viral modulation of programmed necrosis. Curr Opin Virol 2013; 3:296–306; PMID:23773332; http://dx.doi.org/ 10.1016/j.coviro.2013.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khan N, Lawlor KE, Murphy JM, Vince JE. More to life than death: molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr Opin Immunol 2014; 26:76–89; PMID:24556404; http://dx.doi.org/ 10.1016/j.coi.2013.10.017 [DOI] [PubMed] [Google Scholar]
- 92.Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, et al.. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in HARPIN-deficient mice. J Immunol 2014; 192:5476–80; PMID:24821972; http://dx.doi.org/ 10.4049/jimmunol.1400499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al.. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014; 343:1357–60; PMID:24557836; http://dx.doi.org/ 10.1126/science.1249361 [DOI] [PubMed] [Google Scholar]
- 94.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FK, Pasparakis M, Kelliher MA. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 2014; 193:1539–43; PMID:25015821; http://dx.doi.org/ 10.4049/jimmunol.1400590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, Abhyankar A, Israël L, Trevejo-Nunez G, Bogunovic D, et al.. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol 2012; 13:1178–86; PMID:23104095; http://dx.doi.org/ 10.1038/ni.2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang J, Chan FK. Cell biology. RIPK3 takes another deadly turn. Science 2014; 343:1322–3; PMID:24653026; http://dx.doi.org/ 10.1126/science.1252526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE, Vaux DL. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis 2013; 4:e465; PMID:23328672; http://dx.doi.org/ 10.1038/cddis.2012.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, et al.. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis 2014; 5:e1004; PMID:24434512; http://dx.doi.org/ 10.1038/cddis.2013.531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol 2014; 192:2019–26; PMID:24563506; http://dx.doi.org/ 10.4049/jimmunol.1302426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 2010; 7:302–13; PMID:20413098; http://dx.doi.org/ 10.1016/j.chom.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G, et al.. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012; 36:215–27; PMID:22365665; http://dx.doi.org/ 10.1016/j.immuni.2012.01.012 [DOI] [PubMed] [Google Scholar]
- 102.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013; 38:27–40; PMID:23260196; http://dx.doi.org/ 10.1016/j.immuni.2012.09.015 [DOI] [PubMed] [Google Scholar]
- 103.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA, et al.. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A 2014; 111:7391–6; PMID:24799678; http://dx.doi.org/ 10.1073/pnas.1403477111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production. J immunol 2013; 191:5239–46; PMID:24123685; http://dx.doi.org/ 10.4049/jimmunol.1301581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 2014; 192:1835–46; PMID:24453255; http://dx.doi.org/ 10.4049/jimmunol.1302839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, Hu B, Fitzgerald L, Mauldin EA, Copenhaver AM, et al.. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc Natl Acad Sci U S A 2014; 111:7385–90; PMID:24799700; http://dx.doi.org/ 10.1073/pnas.1403252111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sanjo H, Takeda K, Tsujimura T, Ninomiya-Tsuji J, Matsumoto K, Akira S. TAB2 Is Essential for Prevention of Apoptosis in Fetal Liver but Not for Interleukin-1 Signaling. Mol Cell Biol 2003; 23:1231–8; PMID:12556483; http://dx.doi.org/ 10.1128/MCB.23.4.1231-1238.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ali M, Fritsch J, Zigdon H, Pewzner-Jung Y, Schütze S, Futerman AH. Altering the sphingolipid acyl chain composition prevents LPS/GLN-mediated hepatic failure in mice by disrupting TNFR1 internalization. Cell Death Dis 2013; 4:e929; PMID:24263103; http://dx.doi.org/ 10.1038/cddis.2013.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schneider-Brachert W, Tchikov V, Merkel O, Jakob M, Hallas C, Kruse ML, Groitl P, Lehn A, Hildt E, Held-Feindt J, et al.. Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J Clin Invest 2006; 116:2901–13; PMID:17024246; http://dx.doi.org/ 10.1172/JCI23771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, et al.. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 2004; 21:415–28; PMID:15357952; http://dx.doi.org/ 10.1016/j.immuni.2004.08.017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.