

Abstract
Mitogen-activated protein kinase phosphatase 1 (MKP1 or DUSP1) is an antiapoptotic phosphatase that is overexpressed in many cancers, including breast cancer. MKP1 expression is inducible in radiation-treated breast cancer cells, and correlates with human epidermal growth factor receptor 2 (ERBB2, HER2) expression. The role of MKP1 in therapy resistance suggests that targeting MKP1 in HER2-positive breast tumors may significantly enhance the efficacy of anti-HER2 and other anticancer therapies.
Keywords: breast cancer, HER2, mitochondria, MKP1, radioresistance
Abbreviations
- Bcl
B-cell lymphoma
- CD
cluster of differentiation
- ERBB2
v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2
- ERK
extracellular signal regulated kinase
- HER2
human epidermal growth factor receptor 2
- JNK
Jun NH-terminal kinase
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- MKP1
MAPK phosphatase 1
Therapies targeting human epidermal growth factor receptor 2 (HER2, also known as v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 [ERBB2]) are still the leading strategy in the fight against HER2-positive breast cancer. However, a major obstacle to further improving the control rate in patients with breast cancer is the development of resistance in breast cancer cells following chemotherapy and radiotherapy. Understanding the mechanisms of therapy resistance in HER2-positive tumors will enable effective targeting of the specific modulators/mediators of the resistance and will prove beneficial in planning clinical neo-adjuvant therapy. We recently identified a therapy resistance mechanism that functions in breast cancer, and particularly in HER2-positive breast tumors. This mechanism is mediated by a mitogen-activated protein kinase phosphatase, MKP1, via its antiapoptotic activity against Jun NH-terminal kinase (JNK) in the mitochondria (Fig. 1).1
Figure 1.
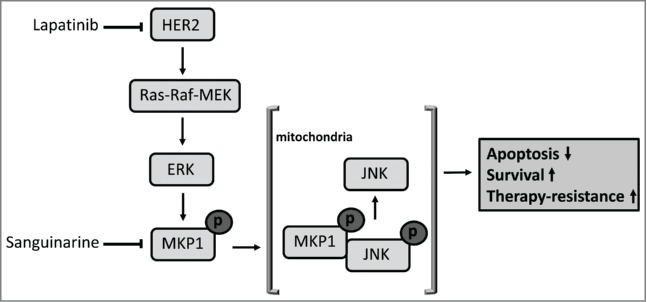
MKP1-mediated survival pathway. HER2-mediated activation of ERK results in the phosphorylation and activation of MKP1. PhosphoMKP1 then translocates into the mitochondria to dephosphorylate and inactivate JNK, resulting in the inhibition of apoptosis and, therefore, enhanced survival. ERK, extracellular-regulated kinase; HER2, human epidermal growth factor receptor 2; JNK, Jun NH-terminal kinase; MKP1, mitogen-activated protein kinase phosphatase 1. Ras–Raf–MEK are mitogen-activated protein kinase signaling cascade proteins: Ras, Rat sarcoma; Raf, rapidly accelerated fibrosarcoma; MEK, MAPK/ERK kinase.
MKP1 (also known as dual specificity phosphatase 1 [DUSP1]) is a threonine-tyrosine dual specificity phosphatase that dephosphorylates and inactivates mitogen-activated protein kinase (MAPKs), extracellular signal regulated kinase (ERK), p38, and JNK in a context-dependent manner.2 Upon radiation, JNK is known to translocate into the mitochondria to initiate mitochondria-mediated apoptosis via its interactions with the Bcl (B-cell lymphoma) family of proteins.3 However, many cancer cells do not show such apoptotic responses after radiation, and the mechanism underlying the non-responsiveness of such tumor cells remains to be elucidated. It is well known that mammalian cells, including tumor cells, are able to generate an adaptive response to genotoxic agents such as ionizing radiation. We had previously shown that in irradiated mouse embryonic fibroblast cells MKP1 specifically targets JNK among the 3 MAP kinases.4 By targeting JNK, MKP1 stops the proapoptotic signals and contributes to cell survival following therapeutic stress. JNK is well known to be overexpressed in breast cancer but, interestingly, its enzymatic activity is decreased in malignant breast tissue.5 These findings led to the discovery of overexpression of MKP1, the upstream phosphatase/deactivator of JNK, in breast cancer. In our recent study, we proposed a mechanism by which therapy resistance occurs in breast cancer via the regulation of mitochondrial JNK by MKP1.1 MKP1 was found to translocate into the mitochondria upon radiation treatment and dephosphorylate mitochondrial JNK to attenuate proapoptotic signals. In doing so, MKP1 provides a survival advantage to breast cancer cells following therapeutic stress. We discovered that mitochondrial translocation of MKP1 is a common mechanism adopted by many mammalian normal and cancer cells in an effort to survive genotoxic stress.
The MAPK signaling cascade starts when an extracellular ligand binds to and activates the family of receptor tyrosine kinases, which includes HER2. The activated receptor then initiates several signaling pathways leading to the activation of MAPKs, ERK, JNK, or p38 MAPK.6 One of the known targets of activated ERK is MKP1, in what is normally a feedback mechanism to stop constitutive signaling from ERK. The fact that ERK is a downstream effector of HER2 signaling indicates how deleterious ERK-mediated activation of MKP1 can be in HER2-overexpressing systems. Continuous activation of MKP1 will result in attenuated JNK proapoptotic activity, leading to increased survival of cancer cells. In our study, intercepting ERK activation via inhibition of either the upstream kinase MEK (MAPK/ERK kinase) or the upstream receptor HER2 resulted in the inhibition of mitochondrial accumulation of MKP1. Consequently, phosphorylated JNK levels were maintained and apoptosis was achieved upon radiation treatment. Notably, we found a correlation between MKP1 and HER2 expression in breast cancer, suggesting that HER2-overexpressing breast cancer may rely on not only the ERK-mediated overactivation of MKP1, but also overexpression of MKP1, for survival. Therefore, our data demonstrated that MKP1 is a key downstream effector of HER2 and may serve as an alternative target for the treatment of cells that are resistant to anti-HER2 therapy.
Another important finding from our laboratory indicated that HER2-expressing breast cancer stem cells (BCSCs; cancer-initiating cells that are ERBB2+/CD44+/CD24−/low) are more radioresistant than HER2-negative BCSCs (ERBB2−/CD44+/CD24−/low; 7). We examined MKP1 expression in HER2-positive and HER2-negative BCSCs and found that MKP1 expression was induced by radiation only in HER2-positive BCSCs. When MKP1 was inhibited via siRNA-mediated knockdown or using the chemical inhibitor sanguinarine, the survival of HER2-positive BCSCs was dramatically reduced following radiation treatment. These results further suggest that, similar to breast cancer cells, BCSCs also depend on MKP1 for survival upon genotoxic stress. The significance of this finding lies in the fact that targeting of cancer-initiating cells will enable the complete eradication of cancer without the risk of recurrence that occurs due to the presence of the inherently therapy-resistant BCSCs within the heterogeneous tumor that are believed to expand to yield resistant metastatic lesions.
Even though targeting MKP1 in a variety of normal and malignant mammalian cells resulted in increased cell death, combinatorial approaches that are designed to interrupt both HER2 signaling and MKP1 activity have proven even more successful. When we treated HER2-overexpressing breast cancer cells and BCSCs with a cocktail of lapatinib (a receptor tyrosine kinase inhibitor) and sanguinarine, we achieved up to a 60% decrease in cell viability. Combined treatment with high-dose radiation and sanguinarine resulted in an approximately 80% decrease in cell survival compared to non-treated breast cancer cells. In future potential clinical trials, MKP1 inhibitors may be considered for adjuvant and/or neo-adjuvant therapy. Therapeutic suppression of MKP activity will enable the proapoptotic benefits of overexpressed JNK in malignant cells and improve the therapy outcome and cancer-free survival of breast cancer patients.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health Grants RO1 CA133402 and RO1 CA152313.
References
- 1.Candas D, Lu CL, Fan M, Chuang FY, Sweeney C, Borowsky AD, and Li JJ. Mitochondrial MKP1 is a target for therapy-resistant HER2-positive breast cancer cells. Cancer Res. 2014; 74(24):7498-509; PMID:25377473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boutros T, Chevet E, and Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev 2008; 60:261-310; PMID:18922965; http://dx.doi.org/ 10.1124/pr.107.00106 [DOI] [PubMed] [Google Scholar]
- 3.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, et al.. Translocation of SAPK/JNK to mitochondria and interaction with Bcl−x(L) in response to DNA damage. J Biol Chem 2000; 275:322-7; PMID:10617621; http://dx.doi.org/ 10.1074/jbc.275.1.322 [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Cao N, Nantajit D, Fan M, Liu Y, Li JJ. Mitogen-activated protein kinase phosphatase-1 represses c-Jun NH2-terminal kinase-mediated apoptosis via NF-kappaB regulation. J Biol Chem 2008; 283:21011-23; PMID:18508759; http://dx.doi.org/ 10.1074/jbc.M802229200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang HY, Cheng Z, Malbon CC. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett 2003; 191:229-37; PMID:12618338; http://dx.doi.org/ 10.1016/S0304-3835(02)00612-2 [DOI] [PubMed] [Google Scholar]
- 6.Sundaresan S, Penuel E, Sliwkowski MX. The biology of human epidermal growth factor receptor 2. Curr Oncol Rep 1999; 1:16-22; PMID:11122793; http://dx.doi.org/ 10.1007/s11912-999-0005-7 [DOI] [PubMed] [Google Scholar]
- 7.Duru N, Fan M, Candas D, Menaa C, Liu HC, Nantajit D, Wen Y, Xiao K, Eldridge A, Chromy BA, et al.. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin Cancer Res 2012. 18:6634-47; PMID:23091114; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1436 [DOI] [PMC free article] [PubMed] [Google Scholar]