

ABSTRACT
Loss of retinoblastoma protein (RB1) function is a major driver in cancer development. We have recently reported that, in addition to its well-documented functions in cell cycle and fate control, RB1 and its paralogs have a novel role in regulating DNA repair by non-homologous end joining (NHEJ). Here we summarize our findings and present mechanistic hypotheses on how RB1 may support the DNA repair process and the therapeutic implications for patients who harbor RB1-negative cancers.
KEYWORDS: DNA repair, non-homologous end-joining, retinoblastoma protein, tumor suppressor
The tumor suppressor retinoblastoma protein (RB1) is a critical component of proliferation and fate control in cells.1,2 Heritable mutations in RB1 cause a highly penetrant predisposition to the childhood cancer retinoblastoma, combined with a substantially increased lifetime risk for other cancers.3 Sporadic mutations are associated with a spectrum of difficult-to-treat cancers including bone and soft tissue sarcoma, small cell lung cancer, breast, endometrial, ovarian, and renal carcinoma.
RB1's functions rely on its interaction with cellular proteins,4 with most known proteins associating with the central A-B pocket (RB1P; amino acid [aa] 379-792) and the RB1C domain (aa 792-928). These include proteins containing a Leu-X-Cys-X-Glu (LXCXE) short linear motif and members of the E2F family of transcription factors, through which RB1 suppresses gene transcription and effects cell cycle control, mitotic fidelity, and cell fate responses (Fig. 1).5-10 Despite evidence for involvement of the third functional domain, RB1N (aa 1-379), in RB1 protein function and tumor suppression,11 the molecular basis through which this domain contributes to these activities has hitherto remained undiscovered. Although the majority of RB1 mutations result in radical protein loss, in both childhood retinoblastoma (http://rb1-lsdb.d-lohmann.de/) and major cancers such as small cell lung cancer and bladder cancer (www.cbioportal.com) some mutations, including small alterations within the RB1N domain, are frame preserving.
Figure 1.
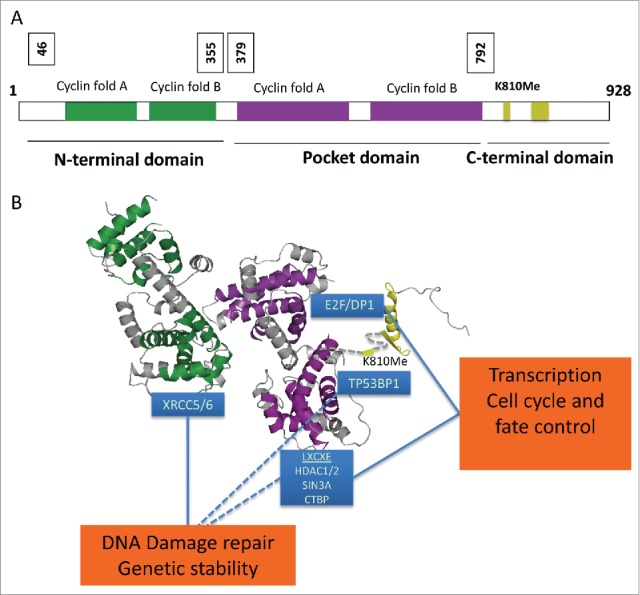
Domains and binding partners of retinoblastoma protein (RB1). (A) Domain organization of RB1, a 928-amino acid protein composed of 3 distinct folded domains, and (B) structural model of RB1 depicting the individual folded domains with annotation of positions of protein binding. Cyclin-like fold pair of the N-terminal domain, RB1N (2QDJ) is colored green, cyclin-like fold pair of the central pocket domain RB1P (3POM) is colored purple, and the C-terminal domain RB1C (2AZE) is colored yellow. The relative positioning of RBN and RBP is described in ref. 24.24 The relative positioning of RBC is inferred by the positioning of the RBP COOH-terminus. Proteins known to bind to distinct regions of RB1 are highlighted in blue boxes. RB1 plays a functional role in transcriptional control of cell proliferation through binding to E2F and Leu-X-Cys-X-Glu (LXCXE) proteins. In addition, RB1 regulates DNA damage repair through the binding of non-homologous end-joining (NHEJ) proteins in the N-terminal domain. By simultaneously binding multiple proteins, such as histone deacetylases (HDACs) via the central pocket domain and the DNA damage repair protein tumor protein p65 binding protein 1 (TP53BP1) via methylated K810 (K810Me) in the C-terminal domain, RB1 may facilitate the recruitment of chromatin regulatory proteins to sites of DNA damage, in turn creating a chromatin environment supportive of DNA damage repair by NHEJ. CTBP, C-terminal binding protein; DP1, DP1 transcription factor; E2F, E2F transcription factor; SIN3A, SIN3 transcriptional regulator family member A; XRCC5/6, X-ray repair complementing defective repair in Chinese hamster cells 5 and 6.
Previous work from our laboratory reporting the crystal structure of RB1N revealed a striking similarity between RB1N and RB1P, predicting an evolutionary relationship and conceptual similarity in function between these 2 RB1 domains.12 Like RB1P, RB1N contains 2 tandem cyclin-like folds, and significant amino acid identity within the corresponding cyclin fold helices suggests that RB1 may have evolved through duplication of an ancestral cyclin fold pair (Fig. 1). Importantly, a surface iso-structural to the ‘cyclin wedge’ through which the cellular cyclins interact with their partner kinase subunits, and which in RB1P is used for the binding of LXCXE proteins,7 is present in RB1N and overt residue conservation within this surface suggests that it is involved in the interaction of RB1N with cellular partner proteins.
Given such structural evidence, together with longstanding documentation that RB1N has the capacity to bind partner proteins in vitro and carries mutations in tumors, we hypothesized that RB1N may mediate tumor suppressor functions through interactions with cellular partner proteins in a similar manner to RB1P. Building on this notion, in our recently published study13 we probed cellular nuclear extracts using affinity purification mass spectrometry. In this proteomic screen, 34 proteins across a range of biologic functions were found to selectively interact with RB1N.13 Among them were several proteins functionally involved in double-strand break DNA repair by non-homologous end joining (NHEJ), namely X-ray repair complementing defective repair in Chinese hamster cells 5 and 6 (XRCC5 and XRCC6), which are required for the recognition of DNA double strand breaks (DSBs), and the double-strand DNA-dependent protein kinase PRKDC (protein kinase, DNA-activated, catalytic polypeptide).14 Structure-guided mutations demonstrated that the cyclin wedge homology surface within RB1N is required for binding of XRCC5 and XRCC6, indicating that this specific RB1N surface, which is predicted to facilitate protein interactions, is responsible for mediating the interaction with NHEJ components.
NHEJ is a prominent pathway in DSB DNA repair and facilitates the rapid joining of broken DNA ends without the need for a homologous repair template.15 NHEJ is critical for the faithful repair of DNA damage during GAP1 (G1) and early S phase of the cell cycle and for the repair of complex DNA damage throughout the cell cycle. Our further functional assessment provides support for the direct functional involvement of RB1 in DNA repair by NHEJ with additional evidence that it shares this function with its paralogs p107 and p130. Notably, 2 RB1 variants that possessed mutations in the cyclin wedge homology region and were therefore unable to physically interact with XRCC6 could not reinstate NHEJ when expressed in RB1-negative cells. Neither of these RB1 variants had a detectable defect in inhibition of cell cycle progression, reduction of colony numbers, or suppression of transcription via E2F, indicating that the roles of RB1 in proliferation control and DNA repair are genetically and functionally separable and independent. We further demonstrate that RB loss adversely affects chromosomal integrity upon irradiation. Specifically, RB loss increased the frequency of chromosomal aberrations in cells independent of mitotic transit, a recognized consequence of defective NHEJ. Together, our data argue that RB1 and its paralogs act to enable NHEJ through physical interaction with components of the canonical NHEJ pathway, and that this activity is unrelated to the canonical cell cycle and transcription regulatory functions of RB1 family proteins.
Outstanding questions remain regarding the mechanism(s) by which the RB1 family proteins support the DNA repair process. It is thought that RB1 orchestrates the assembly of multiprotein complexes in cells through coordinate use of protein binding surfaces. Both RB1P and RB1C interact with chromatin modifiers recognized for their contribution in gene regulation and the regulation of chromatin conformation.10,16 Many of these same modifiers are also known to be involved in NHEJ. These include histone deacetylase 1 and 2 (HDAC 1 and 2) and the histone methyltransferase SUV4 (suppressor of variegation 4-20 homolog 1), which lead to chromatin condensation.17-19 A concurrently published report documents interaction of RB1C with the tudor domain of tumor protein 53 binding protein 1 (TP53BP1),20 which is involved in suppressing the 5′ resection of broken DNA ends, in turn discouraging the use of homology driven repair events (Fig. 1B). Thus, a tempting mechanistic hypothesis would be that RB1N, RB1P, and RB1C enable NHEJ DNA repair in a cooperative manner. In the first stage, RB1 may interact with complexes containing XRCC6 via the RB1N domain, which then may serves as a platform for the subsequent recruitment of chromatin modifiers to the sites of damage by RB1P and RB1C.
Since RB1 loss leads to defects in canonical NHEJ, RB1-negative cancers are likely to be more reliant on other forms of DNA double-strand break repair, including homologous recombination (HR) and alternative inaccurate forms of NHEJ (aNHEJ).21,22 We can thus frame a clinical hypothesis whereby dependencies on alternative repair may identify therapeutic strategies that specifically sensitize cancers with RB1 loss to genotoxic stress. Our findings would predict that inhibition of such repair routes in RB1-defective cancers would offer a therapeutic opportunity by promoting hypersensitivity in these cancers to therapeutic regimens of DNA damage. Loss of RB1 protein function is found in a number of cancers with few options for effective treatment, including small cell lung cancer, ovarian cancer, sarcoma, and triple negative breast cancer. Analogous to the success of poly (ADP-ribose) polymerase (PARP) inhibitors in BRCA1 and BRCA2 (breast cancer 1 and 2, early onset)-deficient patients that display defects in HR-mediated DNA repair,23 future work to characterize the molecular mechanisms of DNA repair in RB1-defective cancers may facilitate the identification of novel therapeutic approaches for patients harboring RB1-negative tumors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The work was supported through Cancer Research UK (grant C107/10433 and CRUK/A15043 to S.M.) and Worldwide Cancer Research (grant number 12-1280 to S.M.). R.C. was supported by an ICR student fellowship. P.H.H. is supported by the Wellcome Trust (WT089028).
References
- 1.Classon M, Harlow E.. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2002; 2:910-7; PMID:12459729; http://dx.doi.org/ 10.1038/nrc950 [DOI] [PubMed] [Google Scholar]
- 2.Khidr L, Chen PL.. RB, the conductor that orchestrates life, death and differentiation. Oncogene 2006; 25:5210-9; PMID:16936739; http://dx.doi.org/ 10.1038/sj.onc.1209612 [DOI] [PubMed] [Google Scholar]
- 3.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP.. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986; 323:643-6; PMID:2877398; http://dx.doi.org/ 10.1038/323643a0 [DOI] [PubMed] [Google Scholar]
- 4.Morris EJ, Dyson NJ.. Retinoblastoma protein partners. Adv Cancer Res 2001; 82:1-54; PMID:11447760 [DOI] [PubMed] [Google Scholar]
- 5.Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D.. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci U S A 1998; 95:10493-8; PMID:9724731; http://dx.doi.org/ 10.1073/pnas.95.18.10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubin SM, Gall AL, Zheng N, Pavletich NP.. Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell 2005; 123:1093-106; PMID:16360038; http://dx.doi.org/ 10.1016/j.cell.2005.09.044 [DOI] [PubMed] [Google Scholar]
- 7.Lee JO, Russo AA, Pavletich NP.. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998; 391:859-65; PMID:9495340; http://dx.doi.org/ 10.1038/36038 [DOI] [PubMed] [Google Scholar]
- 8.Lee C, Chang JH, Lee HS, Cho Y.. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev 2002; 16:3199-212; PMID:12502741; http://dx.doi.org/ 10.1101/gad.1046102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao B, Spencer J, Clements A, Ali-Khan N, Mittnacht S, Broceno C, Burghammer M, Perrakis A, Marmorstein R, Gamblin SJ.. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc Natl Acad Sci U S A 2003; 100:2363-8; PMID:12598654; http://dx.doi.org/ 10.1073/pnas.0436813100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talluri S, Dick FA.. Regulation of transcription and chromatin structure by pRB: here, there and everywhere. Cell Cycle 2012; 11:3189-98; PMID:22895179; http://dx.doi.org/ 10.4161/cc.21263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodrich DW. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. J Cell Physiol 2003; 197:169-80; PMID:14502556; http://dx.doi.org/ 10.1002/jcp.10358 [DOI] [PubMed] [Google Scholar]
- 12.Hassler M, Singh S, Yue WW, Luczynski M, Lakbir R, Sanchez-Sanchez F, Bader T, Pearl LH, Mittnacht S.. Crystal structure of the retinoblastoma protein N domain provides insight into tumor suppression, ligand interaction, and holoprotein architecture. Mol Cell 2007; 28:371-85; PMID:17996702; http://dx.doi.org/ 10.1016/j.molcel.2007.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook R, Zoumpoulidou G, Luczynski MT, Rieger S, Moquet J, Spanswick VJ, Hartley JA, Rothkamm K, Huang PH, Mittnacht S.. Direct Involvement of Retinoblastoma Family Proteins in DNA Repair by Non-homologous End-Joining. Cell Rep 2015; 10:2006-18; PMID:25818292; http://dx.doi.org/ 10.1016/j.celrep.2015.02.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieber MR, Ma Y, Pannicke U, Schwarz K.. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 2003; 4:712-20; PMID:14506474; http://dx.doi.org/ 10.1038/nrm1202 [DOI] [PubMed] [Google Scholar]
- 15.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 2010; 79:181-211; PMID:20192759; http://dx.doi.org/ 10.1146/annurev.biochem.052308.093131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manning AL, Dyson NJ.. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer 2012; 12:220-6; PMID:22318235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai A, Lee JM, Yang WM, DeCaprio JA, Kaelin WG Jr., Seto E, Branton PE.. RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol Cell Biol 1999; 19:6632-41; PMID:10490602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguía R, Dean DC, Esteller M, Jenuwein T, et al.. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 2005; 7:420-8; PMID:15750587; http://dx.doi.org/ 10.1038/ncb1235 [DOI] [PubMed] [Google Scholar]
- 19.Isaac CE, Francis SM, Martens AL, Julian LM, Seifried LA, Erdmann N, Binné UK, Harrington L, Sicinski P, Bérubé NG, et al.. The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol 2006; 26:3659-71; PMID:16612004; http://dx.doi.org/ 10.1128/MCB.26.9.3659-3671.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carr SM, Munro S, Zalmas LP, Fedorov O, Johansson C, Krojer T, Sagum CA, Bedford MT, Oppermann U, La Thangue NB.. Lysine methylation-dependent binding of 53BP1 to the pRb tumor suppressor. Proc Natl Acad Sci U S A 2014; 111:11341-6; PMID:25049398; http://dx.doi.org/ 10.1073/pnas.1403737111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aparicio T, Baer R, Gautier J.. DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst) 2014; 19:169-75; PMID:24746645; http://dx.doi.org/ 10.1016/j.dnarep.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heyer WD, Ehmsen KT, Liu J.. Regulation of homologous recombination in eukaryotes. Annu Rev Genet 2010; 44:113-39; PMID:20690856; http://dx.doi.org/ 10.1146/annurev-genet-051710-150955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lord CJ, Ashworth A.. Targeted therapy for cancer using PARP inhibitors. Curr Opin Pharmacol 2008; 8:363-9; PMID:18644251; http://dx.doi.org/ 10.1016/j.coph.2008.06.016 [DOI] [PubMed] [Google Scholar]
- 24.Lamber EP, Beuron F, Morris EP, Svergun DI, Mittnacht S.. Structural insights into the mechanism of phosphoregulation of the retinoblastoma protein. PLoS One 2013; 8:e58463; PMID:23516486; http://dx.doi.org/ 10.1371/journal.pone.0058463 [DOI] [PMC free article] [PubMed] [Google Scholar]