

ABSTRACT
Runt-related transcription factor 3 (RUNX3) functions downstream of transforming growth factor beta (TGFβ) and plays dual roles in pancreas cancer by both suppressing (by inhibiting proliferation) and promoting (by enhancing migratory and metastatic capacity) disease progression. Consideration of the contextual regulation of RUNX3 together with its myriad downstream effects may help improve clinical outcomes for pancreas cancer patients.
KEYWORDS: Metastasis, pancreatic ductal carcinoma, RUNX3, SMAD4, TGFβ
The last few decades of intense research have not improved the prognosis for patients with pancreatic ductal adenocarcinoma (PDA), which nearly always portends a rapid and painful death. PDA has an unusual proclivity for metastatic spread, with 53% of PDA patients presenting with clinically evident metastatic disease at the time of diagnosis.1 For the remaining 47% of patients with locoregional disease, surgical resection can extend survival but provides little hope for cure.2 Ultimately, these patients also succumb to metastatic or recurrent PDA, suggesting that microscopic dissemination is an early hallmark of the disease.3
Against this relentlessly challenging clinical backdrop, substantial progress has been made toward defining the genetic alterations that contribute to pancreatic cancer initiation and progression. Oncogenic Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations are found in approximately 95% of pancreatic cancer patients and function as an initiating event that is further compounded by additional mutations or loss of tumor suppressor genes such as tumor protein p53 (TP53) and SMAD family member 4 (SMAD4).4 Identification of these cardinal mutations has led to the development of robust preclinical models that faithfully recapitulate the hallmarks of PDA in mice, supplanting less predictive models (such as immortalized cell lines or xenografts) that only partially approximate the phenotypes of autochthonous PDA. As examples, oncogenic Kras and Trp53 mutations have been engineered into their endogenous loci to allow pancreas-specific activation of these alleles using the Cre-Lox system. KrasLSL-G12D/+;Trp53LSL-R172H/+;p48Cre/+ (KPC) mice develop autochthonous tumors of the pancreas that closely mimic the clinical syndrome, histologic features, and metastatic potential of human PDA.5 More recently, a floxed Dpc4/Smad4 allele that allows conditional deletion of this tumor suppressor gene was engineered into KPC mice to generate a KrasLSL-G12D/+;Trp53LSL-R172H/+;Dpc4flox/+;p48Cre/+ (KPDC) model of PDA.6 The primary PDA from these KPDC mice progressed more rapidly than their KPC littermates, at the apparent expense of an attenuated metastatic burden. We identified the transcription factor runt-related transcription factor 3 (Runx3), which is frequently overexpressed in KPC PDA but uncommon in KPDC PDA, as the key factor defining the distinct metastatic potentials of these 2 disease presentations. Runx3 enhanced the migratory potential of invasive KPC PDA cells and also stimulated the release of soluble factors that supported distant colonization of disseminated cells. We further showed an association of RUNX3 expression in the tumor epithelia with patient survival and defined the RUNX3 target osteopontin (SPP1) as a marker for distant relapse in PDA patients who underwent pancreatic resection.
Perhaps not surprisingly for such a potentially important metastatic switch, Runx3 levels are regulated by several inputs operating at both the transcriptional and post-translational levels. In particular, the mutational status of Trp53 and the gene dosage of Dpc4 act cooperatively to define a Runx3 set-point in 3 distinct genetic and phenotypic disease states: (1) highly metastatic and (comparatively) less locally aggressive disease in KPC mice; (2) less metastatic, more locally aggressive PDA in KPDC mice (i.e., loss of one allele of Dpc4); and (3) recovered metastatic potency in a highly proliferative local disease, generating an unusually lethal combination in KPDDC animals (i.e., complete loss in Dpc4/Smad4). Wild-type Trp53 contributes to Runx3 degradation whereas point mutation of Trp53 stabilizes it, leading to elevated levels of Runx3 with loss of heterozygosity (LOH) of Trp53 in KPC animals. Dpc4 gene dosage regulates Runx3 in a biphasic manner: Runx3 levels are high when both copies of Dpc4 are intact, decrease dramatically with loss of one Dpc4 allele, and recover once again with LOH of the locus to generate functionally null Dpc4 tumors. Although the myriad details of the contributors defining Runx3 levels remain to be elucidated, the combined assessment of Runx3 expression and Dpc4 status in primary tumors can potentially be used to predict the most likely cause of patient demise, namely, local disease progression versus distant dissemination, and tailor therapies accordingly.
These findings also contribute to the sometimes vitriolic debate over whether RUNX3 functions as an oncogene or tumor suppressor gene in human malignancies.7,8 We believe it can be either or both, depending not only on the specific context, but also on whether one is considering primary tumor growth or metastatic spread (Fig. 1). In PDA, RUNX3 appears to suppress primary tumor growth through upregulation of key cell cycle inhibitors such as cyclin-dependent kinase inhibitor 1A (CDKN1A or p21), while promoting an invasive and metastatic phenotype by inducing secreted proteins like SPP1 and collagen type VI α1 (COL6A1) that stimulate motility and support distant colonization. This context-dependent classification of RUNX3 as an oncogene or a tumor suppressor gene mirrors, and perhaps contributes to, the dual nature of TGFβ signaling, which lies upstream of RUNX3, in tumorigenesis.9 The biphasic regulation of RUNX3 expression as a function of SMAD4 status further links RUNX3 to the dichotomous TGFβ pathway and also provides a mechanism to promote PDA metastasis in the absence of canonical TGFβ signaling, in which the epithelial-to-mesenchymal transition (EMT) is surprisingly not observed.
Figure 1.
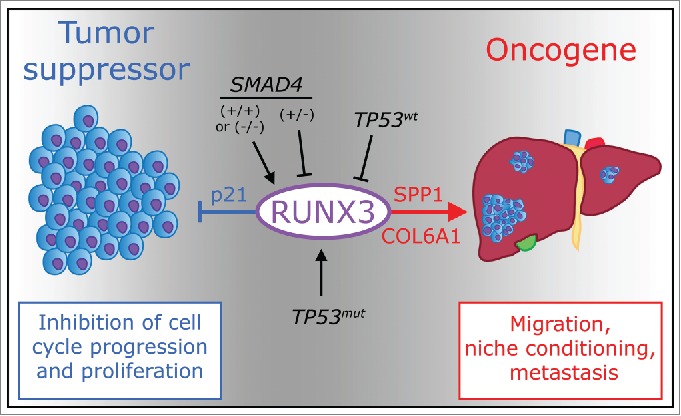
Dual function of RUNX3 as oncosuppressor and oncogene. Expression of runt-related transcription factor 3 (RUNX3) in pancreatic cancer is influenced by genetic alterations of tumor protein p53 (TP53) and SMAD family member 4 (SMAD4). RUNX3 levels respond in a biphasic manner to SMAD4 status: both wild-type and homozygous loss of SMAD4 promote RUNX3 expression, but heterozygous deletion of SMAD4 inhibits it. TP53 mutation and subsequent loss of heterozygosity stabilize RUNX3 expression. RUNX3, in turn, induces expression of cyclin-dependent kinase inhibitor 1A (CDKN1A or p21), inhibiting cell cycle progression and proliferation, and also upregulates both osteopontin (SPP1) and collagen type VI α1 (COL6A1), which promote pancreas cancer cell migration and condition a metastatic niche favoring distant colonization.
Thus, RUNX3 orchestrates a concerted program that tilts the balance from cell division to dissemination and targeting RUNX3 and/or other downstream effectors may help to restrain PDA metastasis. The potential for increased local proliferation with inhibition of RUNX3 can be counterbalanced with complimentary strategies specifically targeting cell cycle mediators such as cyclin-dependent kinases 4 and 6 (CDK4/6).10 As a marker of metastatic potential, however, RUNX3 can also potentially provide a tool to assess the proclivity of a patient's tumor for metastasis versus local growth. This, in turn, can inform the discourse on treatment options for a given patient, maximizing the value of existing modalities such as radiation and cytotoxic chemotherapy, even as we strive to develop more targeted approaches. For example, patients with low RUNX3 levels who are at lower risk for metastatic spread, might be spared the most aggressive systemic treatments (either in the neoadjuvant or adjuvant settings) and may instead benefit from a local therapy such as radiation. Conversely, patients with high RUNX3 in their tumor epithelial cells would be more likely to benefit from systemic chemotherapy, with the possibility of adding radiotherapy depending on the presence or absence of SMAD4. The sequence, scheduling, and duration of the various treatment modalities could likewise be informed by assessing RUNX3 levels and implications for tumor behavior.6 Future studies will be required to validate or refute these hypotheses, but our deepening understanding of the metastatic drive in PDA through such integrated and iterative analyses of human specimens and novel genetically engineered mouse models will ultimately lead to more definitive treatments that significantly change the outlook for patients with this insidious disease.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by National Institutes of Health National Cancer Institute grants CA129537 and CA161112, the Giles W. and Elise G. Mead Foundation and gifts from Maryanne Tagney and David Jones. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.American Cancer Society Cancer Facts & Figures 2015. Atlanta, 2015. [Google Scholar]
- 2.Allison DC, Piantadosi S, Hruban RH, Dooley WC, Fishman EK, Yeo CJ, Lillemoe KD, Pitt HA, Lin P, Cameron JL. DNA content and other factors associated with ten-year survival after resection of pancreatic carcinoma. J Surg Oncol 1998; 67:151-9; PMID:9530884; http://dx.doi.org/ 10.1002/(SICI)1096-9098(199803)67:3%3c151::AID-JSO2%3e3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- 3.Haeno H, Gonen M, Davis MB, Herman JM, Iacobuzio-Donahue CA, Michor F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 2012; 148:362-75; PMID:22265421; http://dx.doi.org/ 10.1016/j.cell.2011.11.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol 2000; 156:1821-5; PMID:10854204; http://dx.doi.org/ 10.1016/S0002-9440(10)65054-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005; 7:469-83; PMID:15894267; http://dx.doi.org/ 10.1016/j.ccr.2005.04.023 [DOI] [PubMed] [Google Scholar]
- 6.Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, Wood LD, Goggins M, Hruban RH, Chang AE, et al.. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 2015; 161:1345-60; PMID:26004068; http://dx.doi.org/ 10.1016/j.cell.2015.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bae SC, Choi JK. Tumor suppressor activity of RUNX3. Oncogene 2004; 23:4336-40; PMID:15156190; http://dx.doi.org/ 10.1038/sj.onc.1207286 [DOI] [PubMed] [Google Scholar]
- 8.Lotem J, Levanon D, Negreanu V, Groner Y. The False Paradigm of RUNX3 Function as Tumor Suppressor in Gastric Cancer. J Cancer Ther 2013; 4:16-25; http://dx.doi.org/ 10.4236/jct.2013.41A003 [DOI] [Google Scholar]
- 9.Chi XZ, Yang JO, Lee KY, Ito K, Sakakura C, Li QL, Kim HR, Cha EJ, Lee YH, Kaneda A, et al.. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/Cip1) expression in cooperation with transforming growth factor {β}-activated SMAD. Mol Cell Biol 2005; 25:8097-107; PMID:16135801; http://dx.doi.org/ 10.1128/MCB.25.18.8097-8107.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011; 11:558-72; PMID:21734724; http://dx.doi.org/ 10.1038/nrc3090 [DOI] [PubMed] [Google Scholar]