

ABSTRACT
The metabolic shift from oxidative phosphorylation to glycolysis as a hallmark of highly aggressive cancer was postulated by Otto Warburg in the 1920s. We identified baculoviral IAP repeat-containing 5 (BIRC5, also known as survivin) as a key player in mitochondrial metabolism and our recent findings suggest glycolysis inhibitors as powerful agents to overcome the antiapoptotic function of survivin in neuroblastoma.
KEYWORDS: Autophagy, glycolysis inhibitors, mitochondria, neuroblastoma, Warburg effect
Baculoviral-IAP-repeat-containing-5 (BIRC5, best known as survivin) was originally identified as a member of the inhibitor of apoptosis (IAP) protein family, but research over the last 20 years has revealed several additional functions of this protein; for example, survivin is involved in mitosis, apoptosis, and autophagy, and is highly expressed during embryonic development. Although it is mostly absent in differentiated adult tissues, recent reports suggest a role for survivin in the survival of hematopoietic, vascular endothelial, and liver cells (reviewed in ref. 11). In transformed cells, however, the expression of survivin is significantly increased, implicating survivin as a prognostic marker for several types of cancer. In neuroblastoma, a childhood malignancy derived from undifferentiated neuronal crest cells, a gain of chromosome 17q causes increased expression of endogenous survivin.2 This genetic defect is associated with increased disease stage, resistance against chemotherapy and radiotherapy, and poor clinical outcome, therefore the development of novel treatment strategies that directly target survivin is a current focus of cancer research.
We recently discovered that survivin not only protects against DNA-damaging agents but also that its expression level defines a threshold for cell death induced by chemotherapeutic agents. In contrast to antiapoptotic X-linked-inhibitor-of-apoptosis-protein (XIAP), for example, this protection occurs upstream of mitochondrial membrane permeabilization,3 suggesting that mitochondrial survivin has a mainly antiapoptotic function.4 However, the underlying molecular mechanism for this protective function against cell death remained enigmatic.
When we overexpressed survivin in neuroblastoma cells, to our surprise the mitochondrial networks changed to punctuate, dot-like structures. These changes in morphology were accompanied by a significant reduction in oxidative phosphorylation (OXPHOS) and a compensatory increase in aerobic glycolysis to cover cellular ATP requirements.5 As a result of the loss of mitochondrial connectivity and the inability to activate respiratory complex I, these cells became highly resistant to forms of cell death that depend on the accumulation of reactive oxygen species (ROS). We further identified the fission protein dynamin-like-protein 1 (DNM1L/Drp1) as responsible for mitochondrial fragmentation in survivin-expressing cells, extending the cellular pathways in which survivin is involved. We therefore concluded that fragmentation of mitochondria via the recruitment of Drp1 by survivin reduces mitochondrial respiration and ROS generation, which mediates chemoresistance in high-stage neuroblastoma.5,6 Additionally, increased glycolysis is accompanied by increased lactate production,7 which fits well into the model of aggressive cancer cells. Importantly, the change in mitochondrial networks and the resulting dependency on glycolysis opens a back door for therapy. We were able to show that neuroblastoma cells carrying a gain of 17q are highly glycolytic and also highly sensitive to glycolysis inhibitors—even low doses of lonidamine (LND) or 2-deoxy-D-glucose (2DG) sensitize these cells to chemotherapeutic agents whereas low-stage neuroblastoma cells and non-transformed cells are fully resistant.5,8 We were able to revert this effect using short-hairpin (sh) RNA, providing evidence that the OXPHOS/glycolysis ratio is regulated by survivin levels.
Detailed analyses on how glycolysis inhibitors target neuroblastoma cells with high survivin expression revealed that survivin is part of a complex that we named the “survivosome” (Fig. 1). Through interaction with beclin 1 (BECN1) and PTEN-induced-kinase-1 (PINK1)–parkin-RBR-E3-ubiquitin-protein-ligase (PARK2, also known as parkin), survivin not only regulates metabolism and cell death inhibition but also defines a threshold for autophagy induction and, interestingly, for its own stability. When cells are treated with 2DG, survivin is rapidly degraded and network-like mitochondria are reformed before the cells undergo death. Since co-treatment with the proteasome inhibitor MG132 and the autophagy inhibitor bafilomycinA1 slows down survivin degradation after 2DG treatment, we speculated that both autophagosomal and proteasomal degradation regulate steady-state levels of survivin. We found that the ubiquitin ligase parkin, which was recently identified as a key player in the elimination of mitochondrial proteins with a basic C-terminus9 (a structural feature also present in survivin), is responsible for survivin degradation and cell death induction after 2DG treatment. Knockdown of either beclin-1 or parkin rescued survivin levels from 2DG-induced degradation and survivin-overexpressing neuroblastoma cells from 2DG-induced death. Glucose withdrawal, on the other hand, induced autophagy independent of survivin levels and cancer stage. The selective effect of 2DG on survivin expression and autophagy-induced cell death was characteristic for neuroblastoma cell lines derived from high-stage tumors with 17q amplification. The 2DG-driven reduction of survivin protein levels apparently results in a feed-forward amplification loop by releasing beclin-1, further promoting autophagy, increasing the loss of survivin, and finally resulting in cell death.8
Figure 1.
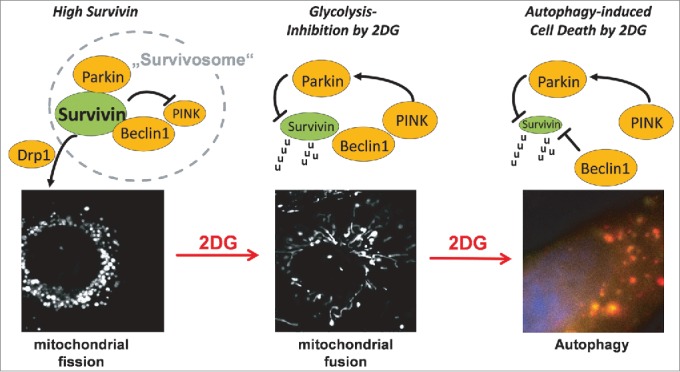
Mitochondrial survivin and glycolysis-inhibition by 2-deoxy-D-glucose (2DG) in neuroblastoma. Increased expression of endogenous survivin causes mitochondrial fission via recruitment of the fission protein Drp1. In such cells mitochondrial survivin interacts with parkin and beclin-1, forming a hypothetical “survivosome” complex. 2DG treatment induces PINK and subsequent phosphorylation/activation of parkin and degradation of survivin, and thereby triggers the formation of mitochondrial networks. The release of beclin-1 from survivin further promotes autophagy-related survivin degradation, autophagosome formation, and autophagic flux, as visualized with a tandem DSRED-LC3-GFP protein. 2DG, 2-deoxy-D-glucose; Drp1, dynamin-like-protein1; GFP, green fluorescent protein; LC3, microtubule-associated protein 1 light chain 3 α (MAP1LC3A); parkin, parkin RBR E3 ubiquitin protein ligase; PINK, PTEN-induced kinase; u, ubiquitin.
Beating the cancer cell at its own game
Targeting survivin in cancer is not a new approach but as survivin is also essential for differentiated somatic cells, it might not be the best one. We chose a different strategy in which we did not neutralize survivin directly but instead targeted the metabolic abnormalities induced by high survivin levels. Thereby, the selective advantage of increased survivin levels in high-stage neuroblastoma, i.e., that these cells escape chemotherapy and may survive under hypoxic conditions, turns into an “Achilles’ heel” for the cancer cells that offers a novel non-genotoxic treatment strategy for children diagnosed with neuroblastoma with gain of 17q. In vivo experiments with tumor cells that show gain of 17q-related survivin overexpression (NB15/shCtr) and NB15 cells in which we reduced survivin overexpression by shRNA (NB15/shSurv) clearly revealed that in neuroblastoma survivin does not primarily contribute to tumor growth, but is mainly involved in protection against cell death. Treatment with 2DG reduced tumor weight and volume to 20% in tumors with endogenously high survivin expression, whereas knockdown of survivin completely abrogated the 2DG-sensitizing effect in NB15/shSurv tumors.8
Therefore, mitochondrial survivin is at the crossroads of mitochondrial structure, respiration, metabolism, cell death, and autophagy. We believe that targeting the survivin-induced Warburg effect may represent a novel non-genotoxic treatment approach for high-stage neuroblastoma.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Mobahat M, Narendran A, Riabowol K. Survivin as a preferential target for cancer therapy. Inter J Mol Sci 2014; 15:2494-516; PMID:24531137; http://dx.doi.org/ 10.3390/ijms15022494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Islam A, Kageyama H, Takada N, Kawamoto T, Takayasu H, Isogai E, Ohira M, Hashizume K, Kobayashi H, Kaneko Y, et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 2000; 19:617-23; PMID:10698506; http://dx.doi.org/ 10.1038/sj.onc.1203358 [DOI] [PubMed] [Google Scholar]
- 3.Obexer P, Hagenbuchner J, Unterkircher T, Sachsenmaier N, Seifarth C, Bock G, Porto V, Geiger K, Ausserlechner M. Repression of BIRC5/survivin by FOXO3/FKHRL1 sensitizes human neuroblastoma cells to DNA damage-induced apoptosis. Mol Biol Cell 2009; 20:2041-8; PMID:19211844; http://dx.doi.org/ 10.1091/mbc.E08-07-0699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, Marusawa H, Zou H, Armstrong R, Matsuzawa S, et al. An IAP-IAP complex inhibits apoptosis. J Biol Chem 2004; 279: 34087-90; PMID:15218035; http://dx.doi.org/ 10.1074/jbc.C400236200 [DOI] [PubMed] [Google Scholar]
- 5.Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2013; 32: 4748-57; PMID:23146905; http://dx.doi.org/ 10.1038/onc.2012.500 [DOI] [PubMed] [Google Scholar]
- 6.Hagenbuchner J, Ausserlechner MJ. Mitochondria and FOXO3: Breath or Die. Frontiers in Physiology 2013; 4; PMID:23801966; http://dx.doi.org/ 10.3389/fphys.2013.00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stern R, Shuster S, Neudecker BA, Formby B. Lactate stimulates fibroblast expression of hyaluronan and CD44: the Warburg effect revisited. Exp Cell Res 2002; 276: 24-31; PMID:11978005; http://dx.doi.org/ 10.1006/excr.2002.5508 [DOI] [PubMed] [Google Scholar]
- 8.Hagenbuchner J, Kiechl-Kohlendorfer U, Obexer P, Ausserlechner MJ. BIRC5/Survivin as a target for glycolysis inhibition in high-stage neuroblastoma. Oncogene 2015; PMID:26148234 [DOI] [PubMed] [Google Scholar]
- 9.Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nat Commun 2013; 4: 1410;PMID:23361001; http://dx.doi.org/ 10.1038/ncomms2400 [DOI] [PubMed] [Google Scholar]