

ABSTRACT
In the past decade mitochondria have emerged as an important cellular signaling hub controlling metabolism, epigenetics, and cell fate. Dysfunctional mitochondria initiate a retrograde nuclear response that influences the cellular reprograming observed in various human pathologies, including cancer. New data suggest that loss of cytochrome c oxidase function promotes the Warburg effect and upregulates several genes with roles in tumor development.
KEYWORDS: Cancer, cytochrome oxidase, mitochondrial dysfunction, retrograde signaling
Nearly 80 years ago Warburg observed that most proliferating cancer cells depend on unusually high glycolytic rates and glucose for energy. He hypothesized that this phenomenon of “aerobic glycolysis” is due to damaged mitochondria of cancer cells. Since then the role of defective mitochondria in cancer development has been intensely debated. Currently, there exist sharply divided views on the possible role of mitochondrial dysfunction in cancer progression.
Mitochondrial dysfunction and retrograde signaling in cancer cells
Mitochondrial function is affected by both genetic and environmental factors. Genetic factors include mitochondrial and nuclear DNA mutations. Mitochondrial DNA (mtDNA) defects such as mutations, deletions, and loss of copy number that result in defective electron transport chain complexes have been reported in a wide variety of cancers.1,2 The causal role of mitochondrial defects in tumor formation has not been established; however, using hybrid cell technology to replace endogenous mtDNA with that from tumor cell lines, the role of mtDNA mutations of Complex I, III, and V subunits in inducing tumorigenic cellular transformations has been demonstrated.2–4 Moreover, defects in nuclear encoded mitochondrial proteins such as succinate dehydrogenase, fumarate hydratase, and isocitrate dehydrogenase have been shown to lead to accumulation of oncometabolites and are directly implicated in specific cancers.2
The mitochondrial and nuclear compartments are functionally linked by signaling networks that regulate the mitochondrial biogenesis and energy production necessary for normal physiologic functions. Pathologic conditions characterized by defective mitochondrial function initiate stress-induced retrograde signaling pathways that are known to alter nuclear gene expression and phenotypic outcomes. Two small-molecule–initiated mechanisms have been extensively researched. One is induced by reactive oxygen species produced by defective mitochondria and was shown to involve pathways mediated by hypoxia inducible factor (Hif-1α), Akt, and MAPK.3,4 The second is triggered by elevated Ca2+ levels in cells with mitochondrial dysfunction and loss of transmembrane potential. This signaling is mediated by calcineurin and calcineurin-activated transcription factors NFkB, C/EBPδ, CREB, and NFAT as signature DNA binding factors and heterogeneous ribonucleoprotein A2 (hnRNPA2) as a transcription co-activator. In many immortalized cells, this pathway has been shown to transform non-tumorigenic cells to a tumorigenic phenotype.5
Role of Cco dysfunction in cellular reprogramming and cancer
Cytochrome c oxidase (CcO) is a key regulator of the electron flux during oxidative phosphorylation. Mutations in mtDNA-coded subunits of CcO have been reported in prostrate, colon, thyroid, and breast cancers.1,2 Although some of these mutations have been functionally characterized,6 the cellular signaling involved and gene targets are largely unknown. Defects in CcO originating from aberrations in nucleus-coded subunits are reported in several pathologic conditions;7 however, their role in cancer development has not been investigated. Among the nuclear-encoded subunits of CcO, subunits IVi1 and Vb have been found to be particularly susceptible to damage under various oxidative stress conditions.7 Our new study examines the nature of signaling initiated by long-term reduction in nuclear CcO subunits and activity and the resulting cellular changes (Fig. 1).8
Figure 1.
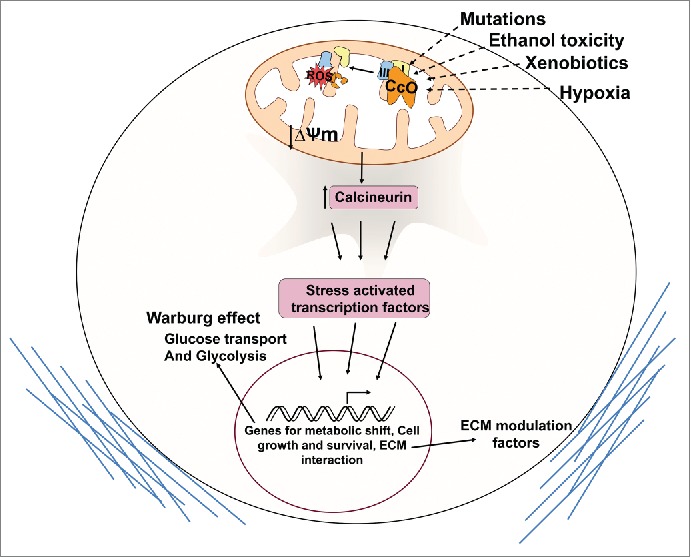
Mitochondrial retrograde signaling induced by cytochrome c oxidase dysfunction promotes the Warburg effect. Cytochrome c oxidase (CcO) defects can be effected by genetic mutations and oxidative damage to susceptible subunits by environmental factors and hypoxia. Retrograde signaling is activated by loss of membrane potential and induction of calcineurin. Stress-induced transcription factors induce gene expression and metabolic reprogramming leading to cell survival and extracellular matrix (ECM) modulation.
In multiple immortalized non-tumor forming cell lines, CcO subunit levels and activity were disrupted by short hairpin RNA (shRNA)-mediated knockdown of either subunit IVi1 or Vb. One of the expected outcomes was a shift in energy production from predominantly oxidative phosphorylation to glycolysis. This was sustained by an increase in glucose uptake and expression of the glucose transporters Glut3 and Glut4. Interestingly Glut1, which is a known target of hypoxia inducible factor (Hif-1α), was unaffected. In concordance with this, although there was a small increase in reactive oxygen species, there was no stabilization of Hif-1α in the CcO subunit knockdown cells. Much like the signaling initiated by mtDNA depletion, loss of CcO activity resulted in increased expression and activation of calcineurin and many of the target genes induced in response to mtDNA depletion. Whole-genome expression analysis in these cell lines surprisingly revealed upregulation of several novel targets. Many of the genes upregulated were secretory factors like TGFβ and proteins that affect the extracellular matrix. Notably, these proteins, such as matrix metalloproteinases, periostin, and tetraspanin, have been shown to play important roles in tumor progression. Interestingly, an earlier study showed that chemical inhibition of CcO activity also upregulated many genes that modulate the extracellular matrix.9 In accordance with these observations, we found that cells with stable knockdown of CcO subunits had a greater potential to invade through Matrigel membrane compared to normal cells, although they had a significantly lower growth rate than normal cells. This finding is in agreement with a recent study that compared bioenergetic parameters of 2 luminal breast cancer cell lines MCF7 and T47D. Interestingly, T47D, which has the higher invasive potential, had a significantly lower CcO IVi1 protein level and reduced mitochondrial efficiency.10 In addition to invasive capacity, CcO-silenced cells formed larger anchorage-independent colonies compared to normal cells. The role of loss of CcO in these changes was verified by ectopic expression of shRNA-resistant CcO subunits, which reversed most of the phenotypic changes.
Future directions
In addition to providing direct evidence in support of mitochondrial dysfunction in cancer as initially proposed by Warburg, our findings strongly support the role of mitochondrial retrograde signaling (MtRS) in human cancer progression. We observed markedly lower levels of CcO subunits in the interior hypoxic core regions of tumor sections of human esophageal squamous cell carcinoma tissues. In light of our findings that many of the proteins upregulated in CcO-deficient cells are secretory and extracellular in nature, it is likely that cells in the hypoxic core produce these factors and influence tumor growth. Another important point to note is the absence of Hif-1α stabilization in these cells, which suggests possible activation of a Hif-1α-independent pathway in some hypoxic tumors. In conclusion, our findings demonstrate the role of MtRS in influencing different aspects of tumor development that might be potential therapeutic targets.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene 2006; 25: 4663-74; PMID:16892080; http://dx.doi.org/ 10.1038/sj.onc.1209604 [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. Mitochondria and cancer. Nat Rev Cancer 2012; 12: 685-98; PMID:23001348; http://dx.doi.org/ 10.1038/nrc3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet 2011; 20: 4605-16; PMID:21890492; http://dx.doi.org/ 10.1093/hmg/ddr395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008; 320: 661-4; PMID:18388260; http://dx.doi.org/ 10.1126/science.1156906 [DOI] [PubMed] [Google Scholar]
- 5.Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell 2004; 14: 1-15; PMID:15068799; http://dx.doi.org/ 10.1016/S1097-2765(04)00179-0 [DOI] [PubMed] [Google Scholar]
- 6.Namslauer I, Brzezinski P. A mitochondrial DNA mutation linked to colon cancer results in proton leaks in cytochrome c oxidase. Proc Natl Acad Sci U S A 2009; 106: 3402-7; PMID:19218458; http://dx.doi.org/ 10.1073/pnas.0811450106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srinivasan S, Avadhani NG. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic Biol Med 2012; 53: 1252-63; PMID:22841758; http://dx.doi.org/ 10.1016/j.freeradbiomed.2012.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srinivasan S, Guha M, Dong DW, Whelan KA, Ruthel G, Uchikado Y, Natsugoe S, Nakagawa H, Avadhani NG. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2015; http://dx.doi.org/ 10.1038/onc.2015.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van WC, Sun Y, Cheung HS, Moraes CT. Oxidative phosphorylation dysfunction modulates expression of extracellular matrix–remodeling genes and invasion. Carcinogenesis 2006; 27: 409-18; PMID:16221732; http://dx.doi.org/ 10.1093/carcin/bgi242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radde BN, Ivanova MM, Mai HX, Salabei JK, Hill BG, Klinge CM. Bioenergetic differences between MCF-7 and T47D breast cancer cells and their regulation by oestradiol and tamoxifen. Biochem J 2015; 465: 49-61; PMID:25279503; http://dx.doi.org/ 10.1042/BJ20131608 [DOI] [PubMed] [Google Scholar]