

ABSTRACT
Glutathione is a major endogenous reducing agent in cells, and cysteine is a limiting factor in glutathione synthesis. Cysteine is obtained by uptake or biosynthesis, and mammalian cells often rely on either one or the other pathway. Because of the scarcity of glutathione, blockade of cysteine uptake causes oxidative cell death known as ferroptosis. A new study suggests that tRNA synthetase suppression activates the endogenous biosynthesis of cysteine, compensates such cysteine loss, and thus makes cells resistant to ferroptosis.
KEYWORDS: Cancer, cysteine, CARS, ferroptosis, methionine, mechanisms of oncogenesis and tumor progression
Defining the connections between metabolic pathways may illuminate therapeutic strategies for treating dysregulated metabolism, tumors, and degenerative disease. Glutathione is a major endogenous reducing agent that protects cells from oxidative stress. Inhibition of glutathione synthesis depletes the glutathione pool in cells; for example, buthionine sulphoximine (BSO) inhibits glutamate-cysteine ligase, the first step in glutathione synthesis, and induces oxidative stress.1 Glutathione is a linear tripeptide, consisting of glutamate, glycine, and cysteine. Of the 3 precursor amino acids, cysteine contributes primarily to the cofactor's reduction potential—its thiol group acts as an electron donor. Glutathione-dependent oxidoreductases, such as glutathione peroxidases (GPXs), transfer 2 electrons from glutathione to effect substrate reduction. Moreover, cysteine is a limiting factor for glutathione biosynthesis and a decrease in cysteine abundance leads to glutathione depletion.
Cysteine can be obtained by mammalian cells in 2 ways. First, the cells can obtain cysteine by importing cystine, the oxidized disulfide of cysteine, via the cystine-glutamate antiporter system xc-.2 Mammalian cells can also synthesize cysteine de novo utilizing 2 amino acids, methionine and serine in a process known as the transsulfuration pathway.3 When cells rely on cystine uptake via system xc- as the primary source of cysteine, inhibition of system xc- causes depletion of cysteine, which subsequently depletes glutathione and can induce oxidative stress and subsequent cell death through a regulated, non-apoptotic form of cell death termed ferroptosis.4,5 This is the lethal mechanism that ensues after pharmacologic inhibition of system xc- by the neurotransmitter glutamate or the small molecule erastin. This process is also relevant to some brain and kidney pathologies.4,5 It has been found that glutathione depletion inactivates glutathione peroxidase 4 (GPX4), a critical cellular antioxidant enzyme that detoxifies lipid hydroperoxides.6 Inhibition of GPX4 enzymatic activity allows accumulation of overwhelming amounts of lipid peroxides, leading to ferroptosis.
Recently, genome-wide siRNA screening for suppressors of ferroptosis revealed that knockdown of cysteinyl-tRNA synthetase, encoded by the CARS gene, inhibits erastin-induced ferroptosis in multiple human and rat cell lines.7 Although erastin- or glutamate-induced lethality was suppressed by CARS knockdown, other classes of ferroptosis-inducing agents, such as inhibitors of glutathione synthesis (e.g., BSO) or GPX4 enzymatic activity, were not suppressed by CARS knockdown (Fig. 1). This indicates that CARS knockdown specifically interferes with ferroptosis induced by cysteine deprivation caused by erastin and glutamate. In fact, metabolomic profiling revealed that CARS knockdown increases cysteine levels in cells. Although the abundance of glutathione itself does not change upon CARS knockdown, the level of cysteinyl glutathione disulfide (CSSG) increases; CSSG may then be reduced to form cysteine and glutathione. Thus, an intriguing hypothesis is that CARS knockdown increases cysteine abundance and thus glutathione synthesis; in addition, excess glutathione may be stored as CSSG, which is reduced to recover glutathione as needed. This view that glutathione abundance is increased by CARS knockdown is supported by the fact that CARS knockdown partially suppressed glutathione depletion upon erastin treatment. Although the regulatory mechanism by which the glutathione level is maintained is not fully understood, this discovery highlights an important new connection between cysteine and glutathione metabolism.
Figure 1.
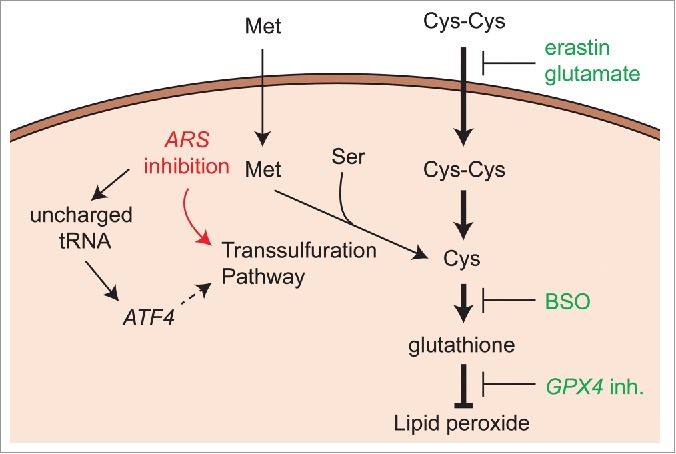
Inhibition of tRNA synthetases restores glutathione from the effects of cystine deprivation in ferroptosis. Ferroptosis-susceptible cells rely on cystine uptake as the primary source of cysteine. However, tRNA synthetase (ARS) inhibition can activate the transsulfuration pathway to synthesize cysteine. The dashed arrow indicates the hypothetical mechanism by which ATF4 activates the transsulfuration pathway through ARS inhibition. Compounds in green indicate ferroptosis inducers targeting different points. ARS, tRNA synthetase; ATF4, activating transcription factor 4; BSO, buthionine sulfoximine; Cys-Cys, cystine; GPX4, glutathione peroxidase 4; Met, methionine; Ser, serine.
A logical question that emerges is how CARS knockdown increases the cysteine pool. CARS knockdown activates de novo cysteine synthesis via upregulation of the transsulfuration pathway (Fig. 1). In fact, not only CARS knockdown, but also inhibition of some other tRNA synthetases (ARS), including histidyl-tRNA synthetases (HARS) or glutamyl prolyl-tRNA synthetases (EPRS), were also shown to activate the transsulfuration pathway and rescue cells from erastin-induced ferroptosis; transcriptional expression of cystathionine-β-synthase (CBS), a rate-limiting enzyme of the pathway,8 was significantly upregulated in response to knockdown of these genes. The suppressive effect of their knockdown on ferroptosis disappeared when the transsulfuration pathway was pharmacologically or genetically inhibited, confirming that ARS knockdown activates the transsulfuration pathway as a mechanism for preventing ferroptosis.
This study raises a number of intriguing questions. First, why does knockdown of some, but not all, ARS activate cysteine synthesis instead of the corresponding amino acids relevant to each synthase? We suspect that ARS suppression that inhibits ferroptosis activates the transcription factor activating transcription factor 4 (ATF4) (Fig. 1), which is activated in response to amino acid deprivation and uncharged tRNAs.9 ARS inhibition increases the presence of uncharged tRNAs, which is a signal for amino acid scarcity, and activates ATF4 expression. ATF4 alters amino acid metabolism pathways, and the transsulfuration pathway is likely one of them.
Second, why is activation of the transsulfuration pathway not the general response to cystine deprivation in all cells? Some cells rely on the transsulfuration pathway as a major supply of cysteine.10 De novo cysteine synthesis induced by CARS knockdown is not sufficient to fully suppress the consequences of glutathione deprivation. Methionine, an essential amino acid and the sole source of sulfur for cysteine biosynthesis, is needed for other biochemical reactions such as methylation; redistributing the metabolic flux of methionine may involve expensive rewiring of metabolic networks. Nonetheless, the finding that loss of CARS suppresses ferroptosis has revealed an unexpected connection between the pathways governing protein synthesis, metabolism and cell death, providing insight into how cells cope with stresses to homeostatic networks.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Miki Hayano for her proofreading and insightful comments on this manuscript. This work was funded by the Howard Hughes Medical Institute, National Institute of Health (5R01CA097061), and New York Stem Cell Science (C026715) to BRS.
References
- 1.Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J Biol Chem 1979; 254:7558-60; PMID:38242. [PubMed] [Google Scholar]
- 2.Jan Lewerenz, Sandra J. Hewett, Ying Huang, Maria Lambros, Peter W. Gout, Peter W. Kalivas, Ann Massie, Ilse Smolders, Axel Methner, Mathias Pergande, Sylvia B. Smith, Vadivel Ganapathy, and Pamela Maher Antioxidants & Redox Signaling. February 10, 2013; 18(5):522-555; http://dx.doi.org/ 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stipanuk MH, Dominy JE, Lee J-I, Coloso RM. Mammalian Cysteine Metabolism: New Insights into Regulation of Cysteine Metabolism. J Nutr 2006; 136:1652S-9S; PMID:16702335. [DOI] [PubMed] [Google Scholar]
- 4.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al.. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012; 149:1060-72; PMID:22632970; http://dx.doi.org/ 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, et al.. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J Am Chem Soc 2014; 136:4551-6; PMID:24592866; http://dx.doi.org/ 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al.. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014; 156:317-31; PMID:24439385; http://dx.doi.org/ 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ 2015; Available from: http://www.nature.com/cdd/journal/vaop/ncurrent/full/cdd201593a.html; PMID:26184909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids 2011; 42:199-205; PMID:21369939; http://dx.doi.org/ 10.1007/s00726-011-0864-8. [DOI] [PubMed] [Google Scholar]
- 9.Palii SS, Kays CE, Deval C, Bruhat A, Fafournoux P, Kilberg MS. Specificity of amino acid regulated gene expression: analysis of genes subjected to either complete or single amino acid deprivation. Amino Acids 2008; 37:79-88; PMID:19009228; http://dx.doi.org/ 10.1007/s00726-008-0199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garg SK, Yan Z, Vitvitsky V, Banerjee R. Differential Dependence on Cysteine from Transsulfuration versus Transport During T Cell Activation. Antioxid Redox Signal 2010; 15:39-47; PMID:20673163; http://dx.doi.org/ 10.1089/ars.2010.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]