

ABSTRACT
ALK (anaplastic lymphoma kinase) inhibitors are approved in for ALK gene rearrangement positive (ALK+) lung cancer, but resistance remains a challenge. We discovered that RAS-RAF-MEK-ERK signaling controls the ALK inhibitor response in ALK+ lung cancer and is critical for ALK inhibitor resistance. Upfront ALK-MEK inhibitor polytherapy may enhance response and forestall resistance.
KEYWORDS: ALK, lung cancer, MEK, resistance, RAS, targeted therapy
The identification of oncogenic alterations that promote cancer growth and the development of oncogene-targeted therapies have revolutionized the treatment of for many cancer patients.1,2 Despite the success of oncogene-targeted therapy, resistance remains a challenge.2 Both primary and acquired drug resistance frequently occur in patients and are often lethal. The molecular determinants of resistance remain unknown in many cases. This is a critical knowledge gap, as understanding of the molecular events that promote innate and acquired resistance would provides biological insight into cell signaling and offers therapeutic strategies to thwart resistance and enhance clinical outcomes.
Our recent work focused on resistance to targeted therapies acting against the oncogenic ALK gene rearrangement (EML4-ALK (; EML4, echinoderm microtubule associated protein like 4) in lung cancers (referred to as ALK+ cancer).3 ALK inhibitors (crizotinib and ceritinib) are initially, yet but only transiently, effective in some, but not all, ALK+ mutant patients because of resistance.4,5 One promising approach to overcome such resistance is the use of a rational upfront combination therapy targeting an oncogenic protein plus a critical downstream pathway. This approach has been successful in BRAFV600E-mutant melanoma patients, in whom upfront targeting of oncogenic BRAF plus its downstream effector MEK yields superior clinical responses when compared to BRAF or MEK inhibitor monotherapy.6 However, which effector pathway is most critical to as a target in cancers with an oncogenic receptor kinase, such as EML4-ALK, is unclear since receptor kinases typically activate multiple pathways.3 We hypothesized that dissection of the pathway dependencies in ALK+ lung cancer cells may reveal the individual pathway that is essential for ALK+ lung cancer cell survival and is the critical determinant of ALK inhibitor response.
To explore this hypothesis, we used a pharmacologic and genetic strategy in which we suppressed or activated the 3 major EML4-ALK effector pathways:, RAF-MEK-ERK (MAPK, mitogen activated protein kinase (MAPK), PI3K (phosphoinositide 3-kinase (PI3K)/AKT, and JAK/STAT (Janus kinase/signal transducer and activators of transcription) signaling.3 We found that RAF-MEK-ERK signaling was uniquely required for ALK+ lung cancer cell survival (Fig. 1);. Whereas whereas inhibition of MEK or ERK was lethal in ALK+ cells, inhibition of PI3K/AKT or JAK/STAT had no effect. We discovered that EML4-ALK engaged all 3 RAS isoforms (H-, N-, K-RAS) to drive RAF-MEK-ERK signaling. This molecular link between EML4-ALK and RAS required the HELP domain in the EML4 component of EML4-ALK, which . This HELP domain in EML4 was essential for proper intracellular localization of EML4-ALK and downstream RAS-RAF-MEK-ERK signaling. We found that ALK+ lung cancer cells and patient tumors that develop resistance to ALK inhibitor treatment reactivate MEK-ERK signaling by 2 mechanisms:. Firstfirst, some tumors harbor copy number gain of wild- type (WT) KRAS and. Secondsecond, patient tumors downregulated the MEK-ERK phosphatase DUSP6, resulting in persistent pathway activation. Both events cause activation of MEK-ERK signaling during ALK inhibitor treatment, thus driving resistance. Inhibition of both MEK and ALK enhanced the initial therapy response and prevented resistance in ALK+ cells, in vitro and in vivo. These data uncover MEK-ERK pathway activation as a hallmark of resistance to ALK targeted therapy and suggest that upfront ALK and MEK co-inhibition may enhance response and patient survival.
Figure 1.
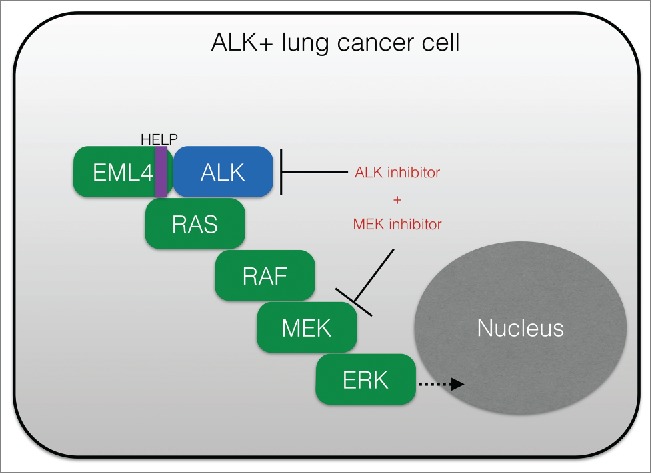
RAS-MAPK signaling promotes cancer cell survival and resistance to ALK-targeting agents. Shown is a schematic of the essential role of RAS-RAF-MEK-ERK (RAS-MAPK) signaling in ALK+ (EML4-ALK positive) tumor cells, and the rational co-targeting strategy to block both ALK and MEK to enhance anti-tumor response. MAPK, mitogen activated protein kinase; ALK, anaplastic lymphoma kinase; HELP, HELP domain in EML4 (echinoderm microtubule associated protein like 4 (EML4).
Open questions
Our findings prompt new avenues for research. First, the findingsy raise the possibility that MEK inhibition may substantially minimize or eliminate resistance in patients with ALK+ lung cancer patients. Efforts are focused on translating the findings into a clinical trial testing ALK inhibitor treatment with a sub-maximal dose of MEK inhibitor in ALK+ patients. The use of a sub-maximal dose of the MEK inhibitor is attractive given the clinical toxicity observed with full-dose MEK inhibition in other patients. Our preclinical data indicate that sub-maximal MEK inhibition is sufficient to enhance the response and eliminate ALK inhibitor resistance, all with improved safety in mice compared to each full-dose monotherapy. Combined ALK plus MEK inhibition may be a viable approach in lung cancer patients, similar to melanoma.6 Yet, the success of this treatment strategy will require careful phase 1 I and pharmacodynamic clinical studies to determine the optimal combination regimen that is safe and effective.
Second, on a fundamental level the findings provide a rationale to investigate the molecular basis of the connection between ALK and RAS-RAF-MEK-ERK signaling. We found that EML4-ALK is expressed on at an intracellular locale that is yet to be fully determined, and that this localization is critical to for engagement of RAS and downstream RAF-MEK-ERK signaling. The Identification of the factors underlying this compartment-specific ALK-RAS regulation and coupling areis critical to understand and a focus of ongoing efforts.
Third, the study reveals the utility value of deciphering pathway dependencies in cancer cells with a particular oncogenic receptor kinase. The development of rational upfront combination therapies will require prioritization, as empiric testing of all possible combination regimens is not possible in patients and could lead to unnecessary toxicity and lack of efficacy. Our approach to prioritize polytherapies may be applied more broadly in cancers with oncogenic receptor kinases such as mutant EGFR (epidermal growth factor receptor) or ROS1 gene rearrangements.7 to define the rational polytherapy mostly likely to maximize clinical responses.
Implications
Our study indicates that RAS-MAPK dependence is a hallmark of ALK+ lung cancers. The data provide a rational basis for upfront testing of a MEK inhibitor (at sub-maximal dose) plus an ALK inhibitor to enhance the magnitude and duration of the anti-tumor response in patients. The findings provide a rationale to explore the cell biologicalular basis of oncogenic receptor kinase signaling, with implications for the design of rational upfront targeted polytherapies in for cancer.
Disclosure of potential conflicts of interest
Dr. Bivona's work has been funded by the NIH (R01, DP2, K08), Howard Hughes Medical Institute, Doris Duke Charitable Foundation, American Lung Association, Sidney Kimmel Foundation for Cancer Research, Searle Scholars Program. He has received compensation as a consultant for Driver Group, Novartis, Clovis Oncology and is a recipient of a research grant from Servier. Dr. Hrustanovic declares no conflict of interest.
References
- 1.Sawyers CL. Lessons learned from the development of kinase inhibitors. Clin Adv Hematol Oncol 2009; 7:588-9; PMID:20020670 [PubMed] [Google Scholar]
- 2.Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov 2012; 2:214-26; PMID:22585993; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0012 [DOI] [PubMed] [Google Scholar]
- 3.Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, et al.. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nature Med 2015; 21(9):1038-47; PMID:26301689; http://dx.doi.org/ 10.1038/nm.3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al.. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Eng J Med 2013; 368:2385-94; PMID:23724913; http://dx.doi.org/ 10.1056/NEJMoa1214886 [DOI] [PubMed] [Google Scholar]
- 5.Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al.. Ceritinib in ALK-rearranged non-small-cell lung cancer. New Eng J Med 2014; 370:1189-97; PMID:24670165; http://dx.doi.org/ 10.1056/NEJMoa1311107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al.. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. New Eng J Med 2014; 371:1877-88; PMID:25265492; http://dx.doi.org/ 10.1056/NEJMoa1406037 [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research N . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511:543-50; PMID:25079552; http://dx.doi.org/ 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]