

ABSTRACT
MDM2 (mouse double minute 2 homolog) and MDMX (double minute X human homolog, also known as MDM4) are critical negative regulators of tumor protein p53. Our recent work shows that MDMX binds to and promotes degradation of retinoblastoma protein (RB) in an MDM2-dependent manner. In a xenograft tumor growth mouse model, silencing of MDMX results in inhibition of p53-deficient tumor growth, which can be effectively reversed by concomitant RB silencing. Thus, MDMX exerts its oncogenic activity via suppression of RB.
KEYWORDS: MDM2, MDMX, p53, RB
Abbreviations
- CDK4
cyclin-dependent protein kinase 4
- E2F
E2F family of DNA binding transcription factors
- MDM2
mouse double minute 2 homolog
- MDMX
double minute X human homolog, also known as MDM4
- p16INK4A
inhibitor of cyclin-dependent kinase type 4
- RB
retinoblastoma protein
- RB1
retinoblastoma susceptibility gene
Inactivation of the retinoblastoma gene is one of the most frequent events in cancer development. The growth suppressive function of the retinoblastoma protein (RB) is inhibited by direct inactivating mutations, or more commonly by changes in expression of its upstream regulators, including overexpression of cyclin D1, activating mutations in cyclin-dependent protein kinase 4 (CDK4), and inactivating mutations in p16INK4A (inhibitor of cyclin-dependent kinase type 4, also called INK4A).1 In addition, RB is inactivated by the viral oncoprotein E7, which promotes RB instability, and by E1A or SV40T, which disrupt the interaction between RB and the E2F family of DNA binding transcription factors (E2F).1
We have shown that mouse double minute 2 homolog (MDM2) directly binds to the C-pocket of RB, leading to disruption of RB–E2F interaction and accelerated RB degradation.2,3 In our recent study, we demonstrated that double minute X human homolog, (MDMX, also known as MDM4) can also bind to and promote RB degradation in an MDM2-dependent manner, thereby promoting tumor growth.4
MDMX and MDM2 are highly homologous with considerable structural similarity.5 Both proteins contain a central acidic domain, a zinc finger domain, and a well-conserved C-terminal typical ring (RING) finger domain, yet only MDM2 possesses E3 ubiquitin ligase activity.5 MDM2 and MDMX are predominantly found as heterodimers, which is thought to enhance E2 protein recruitment and stabilize the ternary complex containing tumor protein p53 (best known as p53).6 In our study, we found that MDMX enhances the ability of MDM2 to bind to and promote RB degradation. Interestingly, although MDM2 binds RB via its central acidic domain,3 MDMX binds RB via its RING finger. Notably, a RING finger mutant MDMX (C463A) retained its ability to bind RB, but did not bind MDM2 and was not able to affect RB protein levels. Hence, MDMX may bring RB and MDM2 into close proximity by recruiting them to its C-terminal site, thus facilitating MDM2 binding to RB and inducing RB degradation. Moreover, MDM2 can promote RB degradation independently of ubiquitination by directly interacting with the C8 subunit of the 20S proteasome to deliver RB for proteolysis.2 It is possible that a MDM2–MDMX heterodimer might more efficiently recruit additional players involved in RB degradation, such as the C8 proteasome subunit, in a manner analogous to E2 binding for p53 ubiquitination. These alternatives are not mutually exclusive, and the molecular mechanism may involve some combination of both possibilities.
As an important alternative pathway to inactivate p53 in addition to p53 gene mutations, amplification of MDM2 or MDMX genes is found in a variety of human tumors and cancers.6 Of note, the MDMX gene is amplified in more than 60% of human retinoblastomas, with an additional 10% of retinoblastomas exhibiting MDM2 gene amplification.7 Notably, retinoblastomas are commonly characterized by a dysfunctional RB pathway but maintain wild-type p53 expression.8 Hence, it is conceivable that loss or inactivation of RB leads to upregulation of E2F transcriptional activity, which in turn promotes apoptosis in a p53-dependent manner.9 Thus, overexpression of MDMX may be selected to inactivate p53 and inhibit p53-mediated apoptosis in the context of RB deficiency in retinoblastoma.
However, numerous cases of cancer have been reported to harbor p53 mutations and concomitant MDMX and/or MDM2 gene amplification.10 An important question then arises as to why the MDMX gene is amplified in cancers where p53 is already inactivated by mutation. Our study indicates that the oncogenic function of MDMX relies on its activity in the suppression of RB. Indeed, silencing of MDMX in p53-null H1299 cells significantly suppressed their growth as xenograft tumors in nude mice. However, concomitant silencing of both MDMX and RB dramatically reverted tumor growth to basal levels, suggesting that MDMX-mediated regulation of RB plays an important role in the pathology of cancer development. These observations suggest that in tumors with wild-type RB1 alleles, overexpression of MDM2 and/or MDMX can facilitate RB degradation and therefore promote cancer development (Fig. 1).
Figure 1.
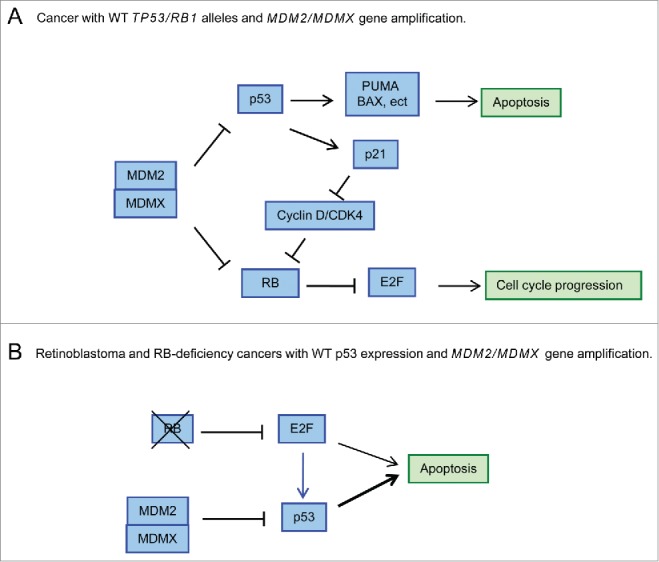
MDM2/MDMX gene amplification in RB wild-type (WT) or RB-deficient cancers with wild-type tumor protein p53. (A) Overexpression of MDM2/MDMX in cancers with WT p53 and RB leads to inactivation of both p53 and RB pathways, resulting in cell proliferation and cancer development. (B) Overexpression of MDM2/MDMX in cancers with WT p53 but RB deficiency inhibits E2F/p53-mediated apoptosis. E2F, E2F family of DNA binding transcription factors; MDM2, mouse double minute 2 homolog; MDMX, double minute X human homolog, also known as MDM4; RB, retinoblastoma protein.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the National Key Basic Research Program (973 Program) of China (2012CB910700) and National Natural Science Foundation of China (NSFC) grant (81330054 and 31171362) to Z-XX, and NSFC grant (31350110216) to JB.
References
- 1.Di Fiore R, D'Anneo A, Tesoriere G, Vento R. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol 2013; 228:1676-87; PMID:23359405; http://dx.doi.org/ 10.1002/jcp.24329 [DOI] [PubMed] [Google Scholar]
- 2.Sdek P, Ying H, Chang DLF, Qiu W, Zheng H, Touitou R, Allday MJ, Xiao ZX. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell 2005; 20:699-708; PMID:16337594; http://dx.doi.org/ 10.1016/j.molcel.2005.10.017 [DOI] [PubMed] [Google Scholar]
- 3.Sdek P, Ying H, Zheng H, Margulis A, Tang X, Tian K, Xiao ZX. The central acidic domain of MDM2 is critical in inhibition of retinoblastoma-mediated suppression of E2F and cell growth. J Biol Chem 2004; 279: 53317-22; PMID:15485814; http://dx.doi.org/ 10.1074/jbc.m406062200 [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Hu L, Qiu W, Deng T, Zhang Y, Bergholz J, Xiao ZX. MDMX exerts its oncogenic activity via suppression of retinoblastoma protein. Oncogene 2015; 34(44):5560-9; PMID:25703327; http://dx.doi.org/ 10.1038/onc.2015.11. [DOI] [PubMed] [Google Scholar]
- 5.Wade M, Li Y-C, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013; 13:83-96; PMID:23303139; http://dx.doi.org/ 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ 2006; 13:927-34; PMID:16543935; http://dx.doi.org/ 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- 7.Laurie NA, Donovan SL, Shih C-S, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, et al.. Inactivation of the p53 pathway in retinoblastoma. Nature 2006; 444:61-6; PMID:17080083; http://dx.doi.org/ 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 8.Kato MV, Shimizu T, Ishizaki K, Kaneko A, Yandell DW, Toguchida J, Sasaki MS. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer Lett 1996; 106:75-82; PMID:8827049; http://dx.doi.org/ 10.1016/0304-3835(96)04305-4 [DOI] [PubMed] [Google Scholar]
- 9.Aslanian A, Iaquinta PJ, Verona R, Lees JA. Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev 2004;18(12):1413-22; PMID:15175242; http://dx.doi.org/ 10.1101/gad.1196704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordon-Cardo C, Latres E, Drobnjak M, Oliva MR, Pollack D, Woodruff JM, Marechal V, Chen J, Brennan MF, Levine AJ. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res 1994; 54:794-9; PMID:8306343. [PubMed] [Google Scholar]