

Abstract
Activation of TLR3 has long been studied in the field of anticancer immunotherapy. Our recent work revealed that TLR3 also promotes the induction of breast cancer stem cells (CSCs) through co-activation of β-catenin and NF-κB signaling. Targeting these 2 pathways simultaneously instead of individually allows for effective inhibition of CSCs that are enhanced by TLR3 activation.
Keywords: β-catenin, breast cancer, cancer stem cells, NF-κB, TLR3
Abbreviations
- CSC
cancer stem cell
- NF-κB
nuclear factor kappa light-chain enhancer of activated B cells
- OCT3/4, octamer-binding transcription factor 3/4, also known as POU5F1 (POU domain, class 5
transcription factor 1)
- NANOG
a transcription factor essential for self-renewal of undifferentiated pluripotent stem cells
- poly(I:C)
polyinosinic-polycytidylic acid
- SOX2
Sex determining region Y-box 2
- TCF
T-cell factor
- TLR
toll-like receptor
Toll-like receptors (TLRs) are key regulators in both the innate and adaptive immune responses. They are highly expressed in antigen-presenting cells and are capable of inducing antitumor mediators such as type I interferon. Accordingly, TLR agonists have been predominantly studied in tumor therapy in attempts to convert immune tolerance into antitumor immunity.1,2
One of 10 TLRs encoded by the human genome, TLR3 is localized in the endosomal compartment and is capable of detecting endogenous macromolecules released by injured tissue and recognizing double-stranded RNA viruses and their synthetic analog polyinosinic-polycytidylic acid [poly(I:C)].1,2 The majority of studies have reported that TLR3 activation leads to tumor suppression.1,2 For example, treatment with the TLR3 agonist poly(I:C) represses tumor growth in mice transplanted with murine prostate cancer cells and reduces the growth of murine melanoma and murine liver tumors. The mechanisms underlying TLR3 activation-induced tumor suppression may be associated with type I interferon secretion, natural killer cell and dendritic cell activation, and/or conversion of tumor-supporting macrophages to tumor suppressors.1
In contrast to the above findings, TLR3 has also been reported to promote tumor development by enhancing cancer cell proliferation and survival.1 Increased TLR3 expression in patients with breast cancer was correlated with poor prognosis.3 Furthermore, the outcomes of clinical trials using TLRs agonists are in general inconsistent.4
It seems that effects of TLR3 (and other TLR) agonists on cancer cells are context-dependent,1 reflecting our insufficient understanding of the direct role of TLR3 in cancer cells, in particular cancer stem cells (CSCs). CSCs have increasingly been considered a key component in cancer recurrence as they are capable of initiating new tumors in vivo and are interconvertible between CSC and non-CSC states (termed CSC plasticity).5 One of the driving forces behind CSC plasticity has been linked to inflammation, although the underlying mechanisms remain poorly understood.
In a recent issue of Cell Death & Differentiation,6 we reported that stimulation of TLR3 promotes breast cancer cells toward a CSC phenotype in vitro and in vivo. Although TLR3 activation induces the death of certain subgroups of tumor cells, it concurrently potentiates the CSC phenotype and tumor-initiating capacity in breast cancer cells. Treatment with the TLR3 agonist poly(I:C) significantly upregulated the expression of CSC markers in both fractionated CSC (CD44high/CD24−/low) and non-CSC (CD44−/low/CD24high) subpopulations. This suggests that an increased pool of CSCs after TLR3 activation may be associated with the proliferation of CSCs together with the induction of a CSC phenotype from non-CSCs.
Additionally, we observed for the first time that TLR3 activation promoted the expression of 3 key pluripotency markers, OCT3/4, NANOG, and SOX2. Notably, overexpressing one of these pluripotency factors has been shown to significantly enhance breast cancer tumorigenesis.7,8 It is possible that TLR3 activation plays a potential role in CSC plasticity and tumor progression. Although the underlying mechanisms remain unclear, one of the driving forces behind CSC plasticity has been closely linked to epigenetic alterations.9 A recent report on adult cell reprogramming suggests that TLR3 stimulation causes rapid and global changes in the expression of epigenetic modifiers to enhance chromatin remodeling and nuclear reprogramming.10 Considering the similarities between cancer stem cells and pluripotent stem cells, it would be interesting to further investigate whether TLR3 enhances the breast CSC phenotype via a mechanism involved in epigenetic alteration.
Moreover, to further confirm the function of TLR3 activation-induced breast CSCs, we carried out secondary xenotransplantation assays. Despite initial growth retardation after TLR3 activation, the acquisition of a CSC phenotype in the remaining tumor cells could engender a stronger and more robust “second wave” of tumor growth. Tumor cells isolated from poly(I:C)-treated mice containing higher numbers of CD44high/CD24−/low cells exhibited a greater than 100-fold higher tumor-initiating capacity than control cells, suggesting a strong tumorigenic potential after poly(I:C) treatment. It can be concluded that TLR3 activation hinders tumor growth initially but enriches for breast CSCs. These observations suggest that the therapeutic potential of a given TLR agonist should be cautiously evaluated with consideration of its possible role in mediating CSC phenotypes and potentiating more robust cancer recurrences.
Within the panoply of molecular players involved in cancer-related inflammation, it is well known that NF-κB is a key orchestrator of the response to TLR activation. Intriguingly, we found that inhibition of NF-κB signaling with either a small molecule or by small interfering RNA knockdown only moderately repressed the breast CSC phenotype induced by TLR3 activation. After examining a number of signaling pathways, we revealed that β-catenin was a co-regulator in the TLR3 activation-enhanced CSC phenotype (Fig. 1). We confirmed this finding using a special β-catenin/T cell factor (TCF)-dependent reporter, although a detailed link between TLR3 and β-catenin/TCF has yet to be defined. Accordingly, knockdown of both NF-κB and β-catenin, but not either one alone, resulted in sufficient repression of TLR3 activation-enriched CSCs. This emphasizes the importance of elucidating co-signaling pathways in CSC evolution for targeted therapy. Given that high expression of TLR3 in breast cancer is correlated with poor clinical prognosis, co-activation of NF-κB and Wnt/β-catenin pathways may be associated with CSC induction and disease relapse, warranting further studies using patient tissue samples. In addition, it would be very interesting to investigate whether, and to what extent, TLR3 activation alters the signaling pathways of tumor stromal cells that constitute the cancer microenvironment and regulate tumor growth.
Figure 1.
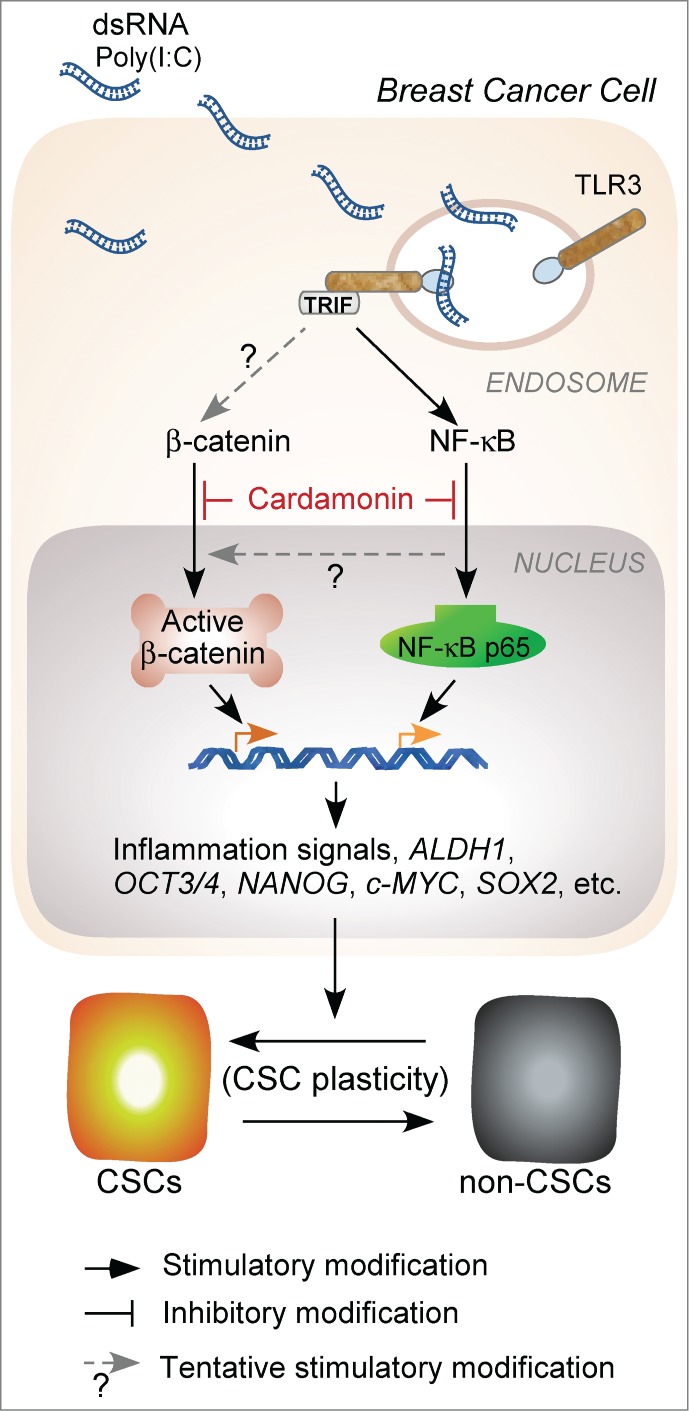
β-catenin signaling is required for breast cancer cells to acquire stem cell features following toll-like receptor 3 (TLR3) activation. Inhibition of both β-catenin and NF-kB is an effective strategy to control the growth of human breast cancer induced by TLR3 activation. c-MYC, NANOG, OCT3/4, and SOX2 are transcriptional factors crucial for the maintenance of pluripotent stem cells and possibly for the induction of CSCs. ALDH1, aldehyde dehydrogenase 1; CSC, cancer stem cell; dsRNA, double-stranded RNA; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NF-κB p65, nuclear factor NF-kB p65 subunit involved in NF-κB heterodimer formation, nuclear translocation, and activation; Poly(I:C), polyinosinic-polycytidylic acid; TRIF, toll/interleukin-1 receptor-domain-containing adapter-inducing interferon-β.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by operating grants from Canadian Breast Cancer Foundation-Ontario Region and the Canadian Institutes of Health Research MOP-111224 to LW.
References
- 1. Pradere JP, Dapito DH, Schwabe RF. The Yin and Yang of Toll-like receptors in cancer. Oncogene 2014; 33(27):3485-95; PMID:23934186; http://dx.doi.org/ 10.1038/onc.2013.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vacchelli E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Toll-like receptor agonists for cancer therapy. Oncoimmunology 2013; 2(8):e25238; PMID:24083080; http://dx.doi.org/ 10.4161/onci.25238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gonzalez-Reyes S, Marin L, Gonzalez L, Gonzalez LO, del Casar JM, Lamelas ML, Gonzalez-Quintana JM, Vizoso FJ. Study of TLR3, TLR4 and TLR9 in breast carcinomas and their association with metastasis. BMC Cancer 2010; 10:665; PMID:21129170; http://dx.doi.org/ 10.1186/1471-2407-10-665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guha M. Anticancer TLR agonists on the ropes. Nat Rev Drug Discov 2012; 11(7):503-5; PMID:22743965; http://dx.doi.org/ 10.1038/nrd3775 [DOI] [PubMed] [Google Scholar]
- 5. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012; 10(6):717-28; PMID:22704512; http://dx.doi.org/ 10.1016/j.stem.2012.05.007 [DOI] [PubMed] [Google Scholar]
- 6. Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S, Chi L, Filion LG, Figeys D, Wang L. β-catenin and NF-κB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ 2015; 22:298-310; PMID:25257174; http://dx.doi.org/ 10.1038/cdd.2014.145, 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vazquez-Martin A, Cufi S, Lopez-Bonet E, Corominas-Faja B, Cuyas E, Vellon L, Iglesias JM, Leis O, Martin AG, Menendez JA. Reprogramming of non-genomic estrogen signaling by the stemness factor SOX2 enhances the tumor-initiating capacity of breast cancer cells. Cell Cycle 2013; 12(22):3471-7; PMID:24107627; http://dx.doi.org/ 10.4161/cc.26692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Noh KH, Kim BW, Song KH, Cho H, Lee YH, Kim JH, Chung JY, Hewitt SM, Seong SY, Mao CP, et al. . Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest 2012; 122(11):4077-93; PMID:23093782; http://dx.doi.org/ 10.1172/JCI64057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 2014; 54(5):716-27; PMID:24905005; http://dx.doi.org/ 10.1016/j.molcel.2014.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES, Pera RR, Yakubov E, Cooke JP. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012; 151(3):547-558; PMID:23101625; http://dx.doi.org/ 10.1016/j.cell.2012.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]