

ABSTRACT
MDM2 is an E3 ubiquitin ligase that binds the N-terminus of p53 and promotes its ubiquitin-dependent degradation. Elevated levels of MDM2 due to overexpression or gene amplification can contribute to tumor development by suppressing p53 activity. Since MDM2 is an oncogene, we explored the possibility that other genetic lesions, namely missense mutations, might alter its activities. We selected mutations in MDM2 that reside in one of the 4 key regions of the protein: p53 binding domain, acidic domain, zinc finger domain, and the RING domain. Unexpectedly, we observed that individual mutations in several of these domains compromised the ability of MDM2 to degrade p53. Mutations in the N-terminal p53 binding domain prevented the formation of a p53–MDM2 complex, thereby protecting p53 from degradation. Additionally, as would be predicted, several cancer-associated mutations in the RING finger domain disrupted the ubiquitin ligase activity of MDM2 and prevented p53 degradation. Interestingly, we observed that amino acid substitutions at the same codon differentially affected MDM2 activity. Our data reveal that mutations in this oncogene can have the paradoxical effect of suppressing its activity. Further understanding of how these mutations perturb MDM2 function may yield novel approaches to inhibiting its activity.
KEYWORDS: Mdm2, ncogenep53, oncogene
Introduction
Mdm2 was first identified as an amplified gene in double-minute chromosomes in transformed mouse fibroblasts.1,2,3 Overexpression or amplification of Mdm2 was subsequently observed in multiple human tumors, including various sarcomas, malignant Schwannomas, gliomas, and leukemias, with a frequency of 7%. It is well established that MDM2 negatively regulates the function of the tumor suppressor p53. MDM2 and p53 are part of a negative feedback loop in which p53 transcriptionally induces MDM2, and MDM2 in turn inactivates p53. Whereas mutation of the TP53 gene is a frequent occurrence in human cancers, tumors that have MDM2 amplification typically retain wild-type p53.4,5 Homozygous deletion of Mdm2 in mice results in lethality at the blastocyst stage due to inappropriate apoptosis driven by elevated p53 activity. This phenotype can be suppressed by depletion of both MDM2 and p53, underscoring the importance of MDM2 in controlling p53 function.6-8 Mdm2 is an E3 ubiquitin ligase that mono- and poly-ubiquitinates p53, resulting in its transport to the cytoplasm and rapid degradation through the 26S proteasome.9,10,11
Human Mdm2 is a 491-amino acid protein possessing a hydrophobic pocket that binds p53 (aa 25–100), a central region with nuclear localization (aa 179–185) and nuclear export signals (aa 179–185), an acidic domain (aa 243–301), and a C-terminus RING (really interesting new gene) domain (aa 432–491).12 The N terminus of Mdm2 forms a deep hydrophobic cleft into which the amphipathic α helix triad of Phe19, Trp23, and Leu26 of p53 fits with steric complementarity. Since Mdm2 binds to the transactivation domain of p53, it can inhibit its transcriptional activity.13,14,15 The intramolecular interaction between the central acidic domain and RING domain of MDM2 is critical for activating and stimulating the catalytic function of the RING domain in promoting ubiquitin release from charged E2.16 The RING domain also harbors a nucleolar localization signal and a nucleotide binding site for adenosine triphosphate (ATP) that regulates its subnuclear distribution.12,17,18 MDM2-mediated cell cycle progression depends upon its interaction with p53 and the RING finger domain.19 However, point mutations within the C-terminus of MDM2 inactivate E3 ligase activity but retain the ability to oligomerize with the wild-type MDM2 RING domain and MdmX. The MDM2–MDM2 interaction requires the central acidic domain and the extreme C-terminal residues of MDM2, whereas MDM2–MDMX interaction requires the proper RING domain structure and the extreme C-terminal residues of MDM2.20,21
Much of what is known about MDM2 activities has been determined from structure/function studies in which different portions of MDM2 have been deleted. Curiously, like its primary target p53, MDM2 is sensitive to subtle changes such as single amino acid substitutions. Site-directed mutagenesis of the N-terminus of MDM2 revealed that residues G58, D68, V75, and C77 are required for the interaction between MDM2 and p53.15 These residues either make direct contact with the p53 protein or have a structural role in MDM2. Similarly, changing a cysteine to an alanine at codon 462 (C462A) in the RING domain of MDM2 inactivates its ubiquitin ligase activity. The C462A MDM2 mutant retains the ability to interact with p53 but is unable to promote its degradation due to the lack of ubiquitin ligase activity. Similar to mice carrying a homozygous deletion of MDM2, mice homozygous for the Mdm2 C462A allele die during embryonic development.22 Further work established that in addition to failing to degrade p53, the C462A MDM2 mutant fails to interact with its binding partner, MDM4.23
In human cancers, MDM2 has been shown to exhibit alternative splicing that eliminates p53 binding domains, the C-terminal RING domain, and the acidic domain.24-26 The protein products of these alternatively spliced forms of MDM2 have been found to be defective in p53 degradation. Likewise, mutations in the MDM2 gene have been found, although analysis of their impact on MDM2 function has been limited. One such cancer-associated mutation that changes cysteine 305 to tyrosine (C305Y) yields a protein that retains the ability to bind and ubiquitinate p53 but fails to promote its degradation because p53 does not translocate from the nucleus to the cytoplasm.27 However, this C305Y MDM2 mutant still binds p53 and inhibits p53 transcriptional activation activity.27
In normal cells the tumor suppressor p53 is maintained at a very low level by Mdm2; this suppression can be inhibited by DNA damage, hypoxia, telomere shortening, and oncogene activation, resulting in increased p53 stability and function.28 More than 50% of human tumors have a mutated form of p53 (mtp53) that is more stable and has gain-of-function activities such as promoting cell growth, chemotherapy resistance, angiogenesis, and metastasis.29,30 An alternative mechanism by which p53 is thought to be inactivated in human cancers is through increased expression of MDM2. To date, there are 92 mutations in the MDM2 gene reported in the Catalog of Somatic Mutations in Cancer (COSMIC) website. Several of these mutations occur in important domains (e.g., the RING domain), suggesting that they might affect MDM2 activities. Since little is known about how mutations in MDM2 can impact its function (i.e., degradation of p53), we analyzed the effect of various cancer-associated mutations. We report that mutations in MDM2 can impair its p53 degrading ability by various mechanisms, including disrupting the MDM2–p53 interaction and inhibiting its ubiquitin ligase activity. Our results indicate that MDM2 can be functionally inactivated with respect to its presumed primary function of degrading p53.
Results
Functional analysis of cancer-associated Mdm2 mutants
One of the primary functions of Mdm2 is to promote the ubiquitin-dependent degradation of p53. To evaluate whether the tumor-associated Mdm2 mutations altered this activity, we performed degradation assays in which we transiently co-transfected the p53 null lung cancer cell line H1299 with wild-type p53 and either wild-type or mutant Mdm2 expression vectors, and then analyzed p53 protein levels as a readout of Mdm2 activity. The effect of the mutations on MDM2 function varied in that some acted like the wild-type protein, some failed to degrade p53, and some exhibited enhanced p53 degradation activity (Fig. 1A-B). The MDM2 mutants A49E, R71Q, R189H, L205Q, V234L, Y287C, W335G, W335L, and M465V were relatively similar to wild-type protein in their ability to degrade p53. Intriguingly, the W335L, S435R, and M465I mutants exhibited an increased capacity to degrade p53 whereas the D86Y, V94M, R189C, C447S, V457A, M465L, and C470Y mutants were essentially unable to degrade p53.
Figure 1.
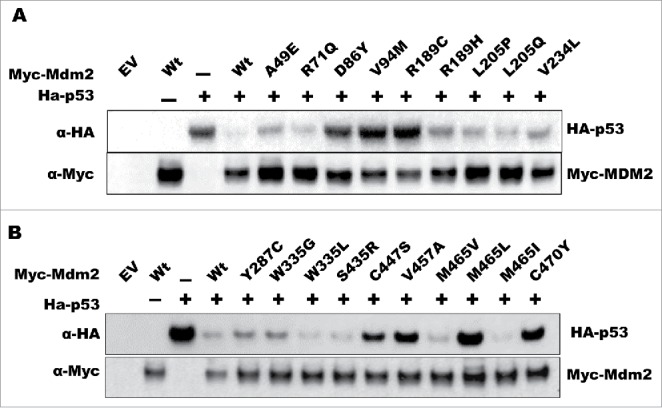
Differential effect of cancer-associated mutations on MDM2 function. H1299 cells were transfected with the indicated plasmids and harvested for western blot analysis the following day. Protein levels were detected using the indicated antibodies.
Analysis of loss-of-function MDM2 mutants
Given the established role of MDM2 as an oncogene in human cancer, we were surprised to see that some of the mutants failed to degrade p53. In the simplest mechanism for MDM2-mediated p53 degradation, there are 2 events that need to occur: (1) MDM2 has to associate with p53, and (2) MDM2 has to promote p53 ubiquitination. Thus, we determined whether the inability of these MDM2 mutants to function properly was due to defects in one of these aspects. We performed a co-immunoprecipitation assay to analyze the interaction between the MDM2 mutants and p53. Compared to wild-type MDM2, 3 mutants—D86Y, V94M, and R189C—exhibited reduced binding to p53 (Fig. 2). In contrast, the other degradation-defective MDM2 mutants bound p53 to the same extent as wild-type MDM2 (Fig. 2). For the D86Y and V94M mutants, the observation of decreased binding is consistent with the location of these 2 amino acid substitutions within the N-terminal domain required for interaction with p53. Since the R189C mutation lies outside of the p53 binding domain, the decreased interaction of this mutant with p53 must be due to a different mechanism.
Figure 2.
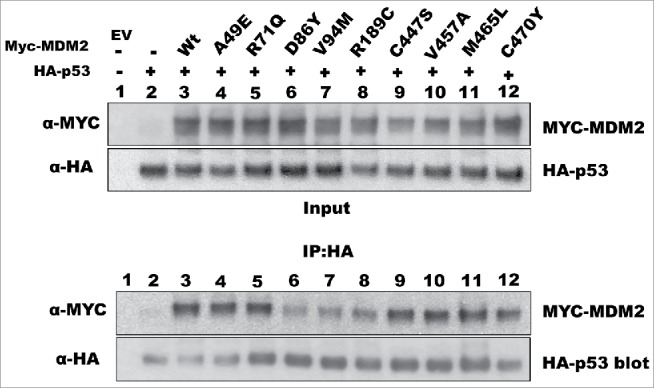
Some MDM2 mutants exhibit decreased interaction with p53. H1299 cells were transfected with the indicated plasmids, lysed on the following day, and processed for immunoprecipitation with an anti-HA antibody (for HA-p53). The association of MDM2 proteins with p53 was detected by western blotting.
The other mutants (C447S, V457A, M465L, and C470Y) interacted with p53 but failed to degrade it (Fig. 2). These mutants have amino acid substitutions that potentially disrupt the RING domain of MDM2, resulting in a loss of function. To test this directly, we examined whether these MDM2 mutants retained ubiquitin ligase activity by comparing their auto-ubiquitination activity with that of the wild-type protein. We co-transfected cells with the MDM2 expression vectors and a HA-tagged ubiquitin vector, treated the cells with MG-132, and immunoprecipitated MDM2 under denaturing conditions to disrupt protein–protein interactions and prevent deubiquitination. Western blot detection of HA-ubiquitin modified MDM2 revealed that the majority of the mutants retained ubiquitin ligase activity (Fig. 3A-B). However, no HA-ubiquitin modification was detected in the C447S, V457A, and C470Y mutants, indicating that these amino acid substitutions inactivate the ubiquitin ligase activity (Fig. 3B). Surprisingly, all the M465 mutants showed active ubiquitin ligase activity despite the fact that the M465L mutant does not degrade p53 (Fig. 1B).
Figure 3.
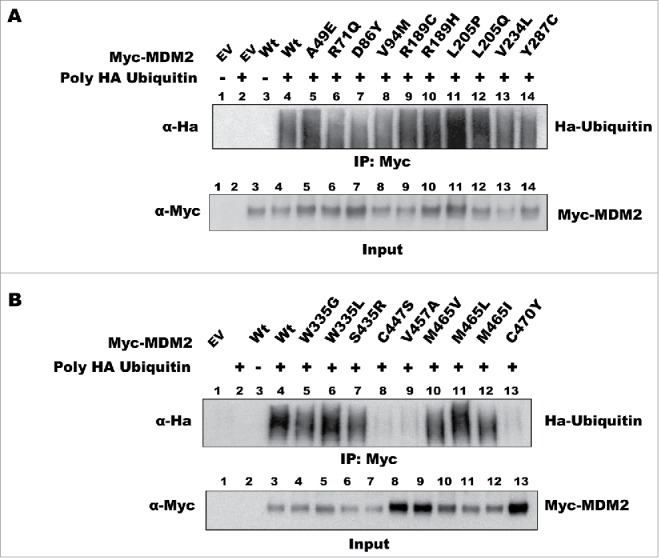
Effect of cancer-associated mutations in the RING domain on MDM2 auto-ubiquitination activity. H1299 cells were transfected with the indicated plasmids. To detect auto-ubiquitination of MDM2, a HA-ubiquitin vector was included in the transfection. The cells were lysed under denaturing conditions and subjected to immunoprecipitation with an anti-myc antibody (to immunoprecipitate myc-tagged MDM2) followed by western blot analysis with an anti-HA antibody to detect the level of ubiquitination. Figs. 3A and 3B show the results from different panels of MDM2 mutants. The immunoprecipitation data are represented as IP: MYC and the corresponding Input blot is labeled accordingly.
Effect of ectopic expression of MDM2 on colony formation
A single nucleotide polymorphism (SNP309G) in the second promoter of the MDM2 gene has been shown to result in elevated expression of the MDM2 transcript and protein.31-34 Mouse embryonic fibroblasts derived from mice carrying humanized MDM2SNP309G alleles express higher levels of MDM2 protein and grow faster than their wild-type counterparts.34 This increased growth rate has been attributed to decreased p53 levels due to high turnover.34 Since some of the MDM2 mutants did not promote p53 degradation, we assessed whether they were still able to increase cell proliferation. We generated stable cell lines of U2OS osteosarcoma cells expressing either wild-type or mutant forms of MDM2 (Fig. 4A). With the exception of the D86Y mutant, we were able to generate cells that ectopically expressed the wild-type or mutant MDM2 proteins (negative data not shown). The inability to generate cells stably expressing the D86Y mutant might be explained by the fact that among the different N-terminal mutants examined, this mutant exhibited the lowest binding to p53. We were surprised to find that cell lines stably expressing the RING finger domain mutants could be generated since they cannot degrade p53 (Fig. 4A). We performed colony formation assays to determine whether ectopic MDM2 expression affected the long-term proliferation rate of these cells. Cells stably expressing wild-type MDM2 generated many more and larger colonies than cells infected with empty vector (Fig. 4B). This indicated that ectopically expressed MDM2 conferred a growth advantage. In contrast, the MDM2 mutants that failed to degrade p53 in our degradation assays generated fewer and smaller colonies than the wild-type MDM2 stable cells (Fig. 4B). Our data indicate that loss of p53 degradation activity of MDM2 reduces the growth promoting properties of this oncogene.
Figure 4.
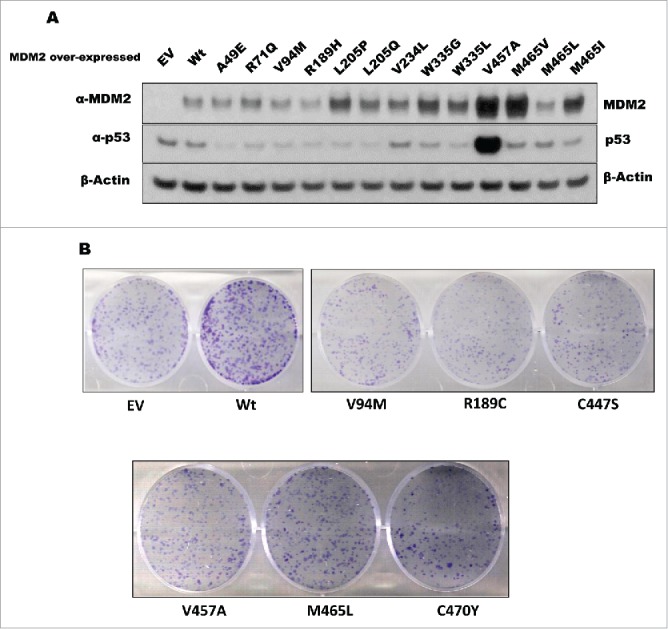
Stable expression of wild-type and mutant MDM2. (A) U2OS cells were transduced with lentivirus carrying empty vector (EV), wild-type MDM2 (Wt), or the indicated MDM2 mutant. After selection, the cells were lysed and subjected to western blot analysis of the indicated proteins. (B) U2OS cells expressing empty vector (EV), wild-type MDM2 (Wt), or MDM2 mutants that exhibited defects in the degradation of p53 were subjected to a colony formation assay to analyze cell growth.
The V457A mutant expressed high levels of p53 due to increased stability of the p53 protein (Supplemental Fig. 1). It was surprising to observe that these cells continued to proliferate despite the fact that they had high levels of p53. In contrast, we were unable to generate stable cell lines with the D86Y mutant, which also failed to degrade p53. However, although these 2 mutants both fail to degrade p53, they differ in that the V457A mutant still interacts with p53 whereas the D86Y mutant does not. We conclude that mutations in MDM2 that disable its interaction with p53 are less well tolerated by cancer cells than mutations that inactivate its ubiquitin ligase activity.
Discussion
We describe the impact of cancer-associated mutations on MDM2 function. Several mutations compromised the ability of MDM2 to degrade p53 through defects in p53 binding or loss of ubiquitin ligase activity. We also observed that mutations in the same residue that differ in the substituted amino acid can have different effects: for example, the R189H mutant was capable of degrading p53 whereas the R189C mutant was not. However, the R189C mutant could interact with p53 and also had ubiquitin ligase activity. Further work is warranted to determine the underlying reason for their differences in biological activity. Another set of mutants at methionine 465 (M465V, M465L, and M465I) were also differentially impacted by the particular amino acid that was substituted. Since the M465L mutant binds p53 and retains ubiquitin ligase activity, we can only speculate that substitution of leucine for methionine at this position disrupts other critical MDM2 interactions. The MDM2 RING domain is required for the formation of heterodimers with MDM4. Since M465L resides within the RING domain, it may be necessary to determine whether the difference between M465L and the other mutants is the ability to interact with MDM4. Further studies are required to understand the nature of the differences between these mutants.
We identified 3 putative gain-of-function MDM2 mutants (W335L, S435R, and M465I) that showed an enhanced ability to promote p53 degradation. We do not know the basis of this increased activity, but nevertheless we can speculate that for one of the mutants (S435R) the missense mutation eliminates a key post-translational modification that negatively regulates MDM2 function. Serine 435 has been shown to be phosphorylated by ATM; this modification inactivates MDM2 by inhibiting its homodimerization and oligomerization.35 Mutation of this residue to aspartic or glutamic acid (i.e., mimicking the negative charge of phosphorylation) reduced the ability of MDM2 to degrade p53, whereas mutation to alanine (i.e. a neutral charge) had no effect.35 Interestingly, we find that substitution of the positively charged amino acid arginine enhanced this activity. Our observation highlights the role of this residue in the regulation of p53 degradation and suggests that it may favor tumor development by reducing p53 stability.
U2OS cells stably overexpressing wild-type MDM2 exhibited increased colony formation ability. In contrast, MDM2 mutants that interacted poorly with p53 or that lacked ubiquitin ligase activity did not enhance U2OS cell proliferation. It is particularly interesting that we were able to stably express MDM2 mutants that had defective ubiquitin ligase activity but not a mutant that failed to interact with p53 (D86Y). Blocking the MDM2–p53 protein–protein interaction has the effect of activating wild-type p53 and has been pursued as a novel anticancer strategy.36,37 A small-molecule inhibitor of the MDM2–p53 interaction, nutlin, is being evaluated in clinical trials. Based on our observation that cells could overexpress ubiquitin ligase-defective MDM2, we speculate that therapeutic approaches that suppress the ubiquitin ligase activity of MDM2 may not be as effective in restoring p53 function as preventing formation of the MDM2–p53 complex. This is supported by the fact that nutlin, which prevents formation of the MDM2–p53 complex, potently suppresses the growth of U2OS cells overexpressing MDM2 (Supplemental Fig. 2). Since MDM2 can also suppress the transcriptional regulatory activity of p53, we speculate that the ubiquitin ligase-defective mutants retain the capacity to suppress p53 function. Our data are in agreement with the recent demonstration that MDM2 knockin mice carrying the Y487A ubiquitin ligase-defective allele progress through embryogenesis and development normally.38 Since sub-lethal stress hyperactivated p53 leading to increased mortality in these mice, this suggests that combined inhibition of MDM2 ubiquitin ligase activity and chemotherapeutic activation of p53 may overcome the antagonism of MDM2. Further studies are required to determine to what extent p53 function is compromised under normal and stressed conditions in cells that express these cancer-associated mutations and to determine whether these mutations sensitize cancer cells to otherwise sub-lethal stress.
In summary, we have characterized how different human cancer-associated mutations affect the degradation of p53 by MDM2. Although p53 is considered the primary target of MDM2, there are other substrates for this E3-ligase; for example, MDM2 has been shown to promote the degradation of human telomerase reverse transcriptase (hTERT), PCNA, Notch4, and Slug.39-43 For 2 of these substrates, Notch4 and Slug, p53 has been reported to be part of a trimeric complex (i.e., Notch4–MDM2–p53 or Slug–MDM2–p53), in which p53 plays a key role in regulating their degradation.41-43 Thus, mutations that abrogate the interaction between p53 and MDM2 could presumably disrupt the formation of these trimeric complexes, resulting in the stabilization of Notch4 and Slug. In a similar manner, mutations in the MDM2 RING domain that disable its ubiquitin ligase activity would retain the capacity to form a trimeric complex between p53 and either Notch4 or Slug, but would be unable to promote their degradation. Thus, these MDM2 mutants might play a role in cancer development by preventing the degradation of proteins that have oncogenic roles. In contrast, the MDM2 mutants that enhance p53 degradation (W335L, S435R, M465I) would promote tumor development by maintaining p53 at low levels. It is worth noting that the SNP309G polymorphism in the MDM2 promoter modestly increases MDM2 mRNA levels (approximately 2 fold) and is associated with an increased risk for development of cancer.31-34 Further in vivo studies using genetically engineered mice carrying one of these point mutations may determine whether, like SNP309G, these mutations can promote tumor development. Taken together, our data provide novel insights into the functional impact of cancer-associated mutations in MDM2 and suggest that these genetic changes in human cancers may not be neutral events.
Material and methods
Plasmid constructs
We have previously described the pCMV-HA-wtp53 vector.29 Human MDM2 cDNA (GE Dharmacon) was amplified by PCR and subcloned into pCMV-Myc (Clontech), which was subsequently used as a template to generate MDM2 point mutants. All mutagenesis was accomplished using the Q5® Site-Directed Mutagenesis Kit from New England BioLabs and appropriate mutagenesis primers were generated using the NEBaseChanger™ tool. Mutations were verified by DNA sequencing.
Cell culture, inhibitors, and transfection
All cell lines used in this study were purchased from the American Tissue Culture Collection (ATCC, Manassas, VA). H1299 (p53 null) cells were cultured in Roswell Park Memorial Institute medium (RPMI-1640) supplemented with 10% FBS and U2OS (p53 positive) osteosarcoma cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS. The inhibitors cycloheximide (CHX) and MG-132 were purchased from Sigma-Aldrich (St. Louis, MO). Transfections were performed using Lipofectamine 2000. Cells were transfected with the indicated plasmids in addition to a CMV-β-gal construct. The cells were lysed and normalized to β-gal activity. To avoid large variations in MDM2 protein levels, we conducted pilot experiments to determine basal expression level and then normalized expression in subsequent experiments.
Generation of stable cell lines
Lentiviruses were generated as previously described.30 U2OS cells were transduced with lentiviral vectors carrying either an empty vector, wild-type MDM2, or MDM2 mutants. The cells were selected for 3 d with 1 μg/mL puromycin and then propagated as polyclonal populations.
In vivo ubiquitination assay
To detect auto-ubiquitination activity of the MDM2 protein, H1299 cells were transfected with plasmids encoding wild-type or mutant Mdm2 together with plasmid expressing HA-tagged ubiquitin as indicated in the text for 20 h and then treated with 20 μM MG-132 for 6 h. Cells were lysed directly in hot 1% sodium dodecyl sulfate (SDS) sample buffer followed by boiling for 5 min at 100°C. The sample buffer was adjusted with stringent buffer as described previously.44 Samples were then incubated with anti-Myc polyclonal antibodies and protein G-agarose beads (KPL, Gaithersburg, Maryland) and rocked for 2 h at 4°C. After addition of sample buffer the beads were boiled for 5 min. MDM2-mediated auto-ubiquitination was assessed by IP with Myc antibody (to detect myc-tagged MDM2) and western blotting with antibody against HA (to detect HA-ubiquitin).
Immunoprecipitation
The interaction between p53 and Mdm2 proteins was detected by transfecting H1299 cells (p53 null) with plasmid constructs PCMV-HA-53 and PCMV-MDM2 wild-type or point mutants together with β-Gal for 24 h, followed by MG-132 treatment for 4 h. Cells were harvested using IP lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.05 mM EDTA, 1% NP40, 10% glycerol) followed by measurement of β-gal activity to normalize transfections. One microgram of mouse MYC monoclonal antibody was added to the samples and rocked for 2 h. Samples were then incubated with Protein G beads for 1 h at 4°C. The beads were washed with IP lysis buffer 5 times, followed by addition of sample loading buffer and boiling for 5 min. Samples were then subjected to standard immunoblotting procedures.
Colony formation assay
Cells were seeded in 6-well plates at a density of 1,000 cells/well. The medium was replenished every 48 h until colonies appeared. After 10 d the medium was removed, the plates were washed with PBS, and 1 mL of staining solution (0.5% crystal violet and 6% glutaraldehyde) was added and incubated for 30 min. The plates were immersed in DI water to remove excess stain and the stained colonies were counted.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the grant NCI CA166974–01A1 to L.A.M.
Reference
- 1.Cahilly-Snyder L, Yang-Feng T, Francke U, George DL. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somatic Cell Mol Genetics 1987; 13:235-44; PMID:13474784; http://dx.doi.org/2026149 10.1007/BF01535205 [DOI] [PubMed] [Google Scholar]
- 2.Fakharzadeh S, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J 1991; 10:1565; PMID:2026149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Küpper M, Joos S, Von Bonin F, Daus H, Pfreundschuh M, Lichter P, Trümper L. MDM2 gene amplification and lack of p53 point mutations in Hodgkin and Reed–Sternberg cells: results from single‐cell polymerase chain reaction and molecular cytogenetic studies. British J Haematol 2001; 112:768-75; PMID:11260082; http://dx.doi.org/9671804 10.1046/j.1365-2141.2001.02566.x [DOI] [PubMed] [Google Scholar]
- 4.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. 1993; 362(6423):857-60 [DOI] [PubMed] [Google Scholar]
- 5.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res 1998; 26:3453-9; PMID:9671804; http://dx.doi.org/ 10.1093/nar/26.15.3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Oca Luna RM, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378:203-6; PMID:7477326; http://dx.doi.org/ 10.1038/378203a0 [DOI] [PubMed] [Google Scholar]
- 7.Steinman HA, Jones SN. Generation of an Mdm2 conditional allele in mice. Genesis 2002; 32:142-4; PMID:11857802; http://dx.doi.org/ 10.1002/gene.10035 [DOI] [PubMed] [Google Scholar]
- 8.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378:206-8; PMID:7477327; http://dx.doi.org/ 10.1038/378206a0 [DOI] [PubMed] [Google Scholar]
- 9.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387:296-9; PMID:9153395; http://dx.doi.org/ 10.1038/387296a0 [DOI] [PubMed] [Google Scholar]
- 10.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J 2000; 19:94-102; PMID:10619848; http://dx.doi.org/ 10.1093/emboj/19.1.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letters 1997; 420:25-7; PMID:9450543; http://dx.doi.org/ 10.1016/S0014-5793(97)01480-4 [DOI] [PubMed] [Google Scholar]
- 12.Fåhraeus R, Olivares-Illana V. MDM2s social network. Oncogene 2013; 33(35):4365-76; PMID:24096477; http://dx.doi.org/7686617 10.1038/onc.2013.410 [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 1993; 13:4107-14; PMID:7686617; http://dx.doi.org/ 10.1128/MCB.13.7.4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996; 274:948-53; PMID:8875929; http://dx.doi.org/ 10.1126/science.274.5289.948 [DOI] [PubMed] [Google Scholar]
- 15.Freedman DA, Epstein CB, Roth JC, Levine AJ. A genetic approach to mapping the p53 binding site in the MDM2 protein. Mol Med 1997; 3:248; PMID:9131587 [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Q, Song T, Chen L, Chen J. Autoactivation of the MDM2 E3 ligase by intramolecular interaction. Mol Cell Biol 2014; 34:2800-10; PMID:24842904; http://dx.doi.org/ 10.1128/MCB.00246-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Priest C, Prives C, Poyurovsky MV. Deconstructing nucleotide binding activity of the Mdm2 RING domain. Nucleic Acids Res 2010; 38:7587-98; PMID:20671028; http://dx.doi.org/ 10.1093/nar/gkq669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poyurovsky MV, Jacq X, Ma C, Karni-Schmidt O, Parker PJ, Chalfie M, Manley JL, Prives C. Nucleotide binding by the Mdm2 RING domain facilitates Arf-independent Mdm2 nucleolar localization. Mol Cell 2003; 12:875-87; PMID:14580339; http://dx.doi.org/ 10.1016/S1097-2765(03)00400-3 [DOI] [PubMed] [Google Scholar]
- 19.Argentini M, Barboule N, Wasylyk B. The contribution of the RING finger domain of MDM2 to cell cycle progression. Oncogene 2000; 19:3849-57; PMID:10951578; http://dx.doi.org/ 10.1038/sj.onc.1203737 [DOI] [PubMed] [Google Scholar]
- 20.Leslie PL, Ke H, Zhang Y. The MDM2 RING Domain and Central Acidic Domain Play Distinct Roles in MDM2 Protein Homodimerization and MDM2-MDMX Protein Heterodimerization. J Biol Chem 2015; 290:12941-50; PMID:25809483; http://dx.doi.org/ 10.1074/jbc.M115.644435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C‐terminus of MDM2 can be provided by MDMX. EMBO J 2007; 26:102-12; PMID:17159902; http://dx.doi.org/ 10.1038/sj.emboj.7601469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindström MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 2007; 12:355-66; PMID:17936560; http://dx.doi.org/ 10.1016/j.ccr.2007.09.007 [DOI] [PubMed] [Google Scholar]
- 23.Clegg HV, Itahana Y, Itahana K, Ramalingam S, Zhang Y. Mdm2 RING mutation enhances p53 transcriptional activity and p53-p300 interaction. PLoS One 2012; 7:e38212; PMID:22666487; http://dx.doi.org/ 10.1371/journal.pone.0038212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans SC, Viswanathan M, Grier JD, Narayana M, El-Naggar AK, Lozano G. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene 2001; 20:4041-9; PMID:11494132; http://dx.doi.org/ 10.1038/sj.onc.1204533 [DOI] [PubMed] [Google Scholar]
- 25.Sigalas I, Calvert AH, Anderson JJ, Neal DE, Lunec J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: transforming ability and frequent detection in human cancer. Nat Med 1996; 2:912-7; PMID:8705862; http://dx.doi.org/ 10.1038/nm0896-912 [DOI] [PubMed] [Google Scholar]
- 26.Bartel F, Taubert H, Harris LC. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002; 2:9-15; PMID:12150820; http://dx.doi.org/ 10.1016/S1535-6108(02)00091-0 [DOI] [PubMed] [Google Scholar]
- 27.Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol Cell Biol 2007; 27:1056-68; PMID:17116689; http://dx.doi.org/ 10.1128/MCB.01307-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pant V, Lozano G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev 2014; 28:1739-51; PMID:25128494; http://dx.doi.org/ 10.1101/gad.247452.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, Chauhan K, Daniels SR, Boccetta M, Garrett MR. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev 2012; 26:830-45; PMID:22508727; http://dx.doi.org/ 10.1101/gad.181685.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, Carrero ZI, Ramakrishnan G, Watabe K, Haupt Y. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nature Commun 2015; http://dx.doi.org/ 10.1038/ncomms8389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, Bartel F, Taubert H, Wuerl P, Hait W, et al.. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res 2006; 66:5104-10; PMID:16707433; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-0180 [DOI] [PubMed] [Google Scholar]
- 32.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al.. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004; 119:591-602; PMID:15550242; http://dx.doi.org/ 10.1016/j.cell.2004.11.022 [DOI] [PubMed] [Google Scholar]
- 33.Bond GL, Menin C, Bertorelle R, Alhopuro P, Aaltonen LA, Levine AJ. MDM2 SNP309 accelerates colorectal tumour formation in women. J Med Genet 2006; 43:950-2; PMID:16825430; http://dx.doi.org/ 10.1136/jmg.2006.043539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Post SM, Quintas-Cardama A, Pant V, Iwakuma T, Hamir A, Jackson JG, Maccio DR, Bond GL, Johnson DG, Levine AJ, et al.. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell 2010; 18:220-30; PMID:20832750; http://dx.doi.org/ 10.1016/j.ccr.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J 2009; 28:3857-67; PMID:19816404; http://dx.doi.org/ 10.1038/emboj.2009.294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med 2007; 13:23-31; PMID:17126603; http://dx.doi.org/ 10.1016/j.molmed.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 37.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al.. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844-8; PMID:14704432; http://dx.doi.org/ 10.1126/science.1092472 [DOI] [PubMed] [Google Scholar]
- 38.Tollini LA, Jin A, Park J, Zhang Y. Regulation of p53 by Mdm2 E3 ligase function is dispensable in embryogenesis and development, but essential in response to DNA damage. Cancer Cell 2014; 26:235-47; PMID:25117711; http://dx.doi.org/ 10.1016/j.ccr.2014.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh W, Lee EW, Lee D, Yang MR, Ko A, Yoon CH, Lee HW, Bae YS, Choi CY, Song J. Hdm2 negatively regulates telomerase activity by functioning as an E3 ligase of hTERT. Oncogene 2010; 29:4101-12; PMID:20453884; http://dx.doi.org/ 10.1038/onc.2010.160 [DOI] [PubMed] [Google Scholar]
- 40.Groehler AL, Lannigan DA. A chromatin-bound kinase, ERK8, protects genomic integrity by inhibiting HDM2-mediated degradation of the DNA clamp PCNA. J Cell Biol 2010; 190:575-86; PMID:20733054; http://dx.doi.org/ 10.1083/jcb.201002124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Bae S, An S, Park JK, Kim EM, Hwang SG, Kim WJ, Um HD. Cooperative actions of p21WAF1 and p53 induce Slug protein degradation and suppress cell invasion. EMBO Rep 2014; 15:1062-8; PMID:25141863; http://dx.doi.org/ 10.15252/embr.201438587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, et al.. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol 2009; 11:694-704; PMID:19448627; http://dx.doi.org/ 10.1038/ncb1875 [DOI] [PubMed] [Google Scholar]
- 43.Sun Y, Klauzinska M, Lake RJ, Lee JM, Santopietro S, Raafat A, Salomon D, Callahan R, Artavanis-Tsakonas S. Trp53 regulates Notch 4 signaling through Mdm2. J Cell Sci 2011; 124:1067-76; PMID:21402876; http://dx.doi.org/ 10.1242/jcs.068965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Govers R, van Kerkhof P, Schwartz AL, Strous GJ. Linkage of the ubiquitin‐conjugating system and the endocytic pathway in ligand‐induced internalization of the growth hormone receptor. EMBO J 1997; 16(16):4851-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.