

Abstract
The tumor suppressor Smad4/DPC4 is an essential transcription factor in the TGF-β pathway that was previously thought to function constitutively. We recently reported that Smad4 activity and stability are directly regulated by 2 major signaling pathways, RTK/MAPK and Wnt/GSK3. Here we examine the molecular, cellular, and potential therapeutic significance of these findings.
Keywords: EGF, FGF, GSK3, TGF-β, Ras, Erk, Smad, tumor suppressor, Wnt, β-TrCP
The transforming growth factor-β (TGF-β) family of growth factors controls a broad spectrum of biological processes such as cell differentiation, proliferation, migration, apoptosis, and extracellular matrix remodeling.1 Despite these multiple roles, the canonical TGF-β signaling pathway is surprisingly simple: TGF-β receptors signal by phosphorylating C-terminal serine residues of the transcription factors Smad1/5/8 for bone morphogenetic proteins (BMPs) or pSmad2/3 for the TGF-β/activin branch of the pathway.1 The transcription factor Smad4 functions as a co-Smad that binds to receptor-phosphorylated Smads and was, until recently, considered an unregulated and constitutively active component of the pathway.
Glycogen synthase kinase-3 (GSK3) is a Wnt- and phosphoinositide 3-kinase (PI3K)-regulated kinase that requires pre-phosphorylated substrates. For phosphorylation of a serine/threonine (Ser/Thr) residue, GSK3 prefers a phospho-Ser/Thr at the n + 4 position, known as the priming phosphorylation site.2 During a bioinformatics screen of the human proteome,3 we noticed that Smad4 contains 3 putative threonine GSK3 phosphorylation sites primed by a canonical mitogen-activated protein kinases (MAPK) site (PxTP), prompting us to investigate the biological functions of these phosphorylations. Using a custom-made phospho-specific antibody, we found that Smad4 is indeed phosphorylated by GSK3 after treatment with fibroblast growth factor (FGF).4
FGF stimulation leads to phosphorylation of Smad4 by Erk at the canonical MAPK site located at threonine 277. This phosphorylation event has a dual effect on Smad4 activity.4 First, it allows Smad4 to reach its peak of transcriptional activity by activating a growth factor-regulated transcription activation domain located in the Smad4 linker region (Fig. 1A). Second, MAPK primes Smad4 for GSK3-mediated phosphorylations that cause transcriptional inhibition and also generate a phosphodegron that is used as a docking site by the ubiquitin E3 ligase beta-transducin repeat containing (β-TrCP) (Fig. 1A). We found that Wnt, which triggers the sequestration of GSK3 inside multivesicular bodies,3 inhibited Smad4 phosphorylation by GSK3 and potentiated the TGF-β signal; this stimulatory effect of Wnt was particularly striking at low levels of TGF-β ligand.4 Importantly, Wnt increases TGF-β signaling only when FGF (or epidermal growth factor [EGF], depending on the cell line) is present. In the absence of the priming MAPK phosphorylation, the Wnt and TGF-β signaling pathways remain entirely insulated from each other.4 These findings provide a novel mechanism for integrating 3 signaling pathways that would otherwise remain insulated, allowing cells to sense the activation status of the MAPK and Wnt pathways and adapt the TGF-β outcome to their cellular context.
Figure 1.
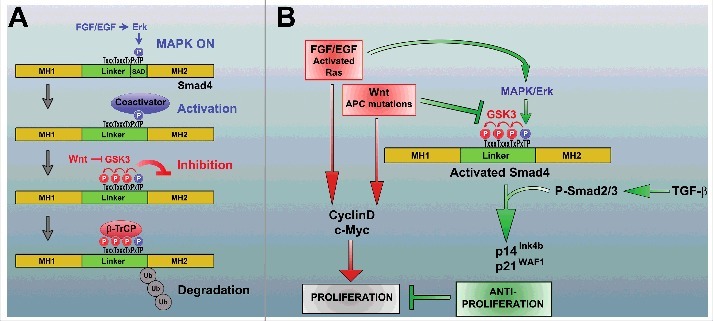
Regulation of Smad4 by the FGF/MAPK and Wnt signaling pathways. (A) Fibroblast growth factor (FGF) stimulates Smad4 phosphorylation by Erk at Thr 277. This has a dual function. First, it allows Smad4 to reach peak transcriptional activity (the identity of the co-activator is still unknown). Second, Erk/mitogen-activated protein kinase (MAPK) phosphorylation primes Smad4 for glycogen synthase kinase-3 (GSK3) phosphorylations that cause transcriptional inhibition and also generate a phosphodegron that serves as a docking site for the ubiquitin E3 ligase beta-transducin repeat containing protein (β-TrCP). Thus, both the activity and the stability of Smad4 are regulated by the FGF/EGF and Wnt signaling pathways. (B) Model highlighting Smad4 regulation in tumor suppression. Many tumors harbor activating Ras/Raf mutations that activate the Erk pathway, thus stimulating proliferation (red arrows). When the Wnt pathway is also activated (for example by loss of adenomatous polyposis coli [APC]), the activity and stability of Smad4 will be enhanced through the newly described regulatory mechanism (green arrows). This gain in Smad4 activity increases the antiproliferative effects of TGF-β (by increasing transcription of cyclin-dependent kinase (CDK) inhibitors such as p14Ink4b and p21WAF1), counteracting the proliferation driven by the Ras and Wnt pathways (red arrows). When Smad4 function is mutated during cancer progression, the barrier effect of Smad4 is lost with catastrophic consequences.
Smad4 is also known as deleted in pancreatic carcinoma 4 (DPC4), and acts as a major tumor suppressor gene that constrains cancer growth. Pancreatic, colorectal, and prostate carcinomas proliferate rapidly and progress toward metastases when Smad4 function is lost.5–7 At early stages, many tumors are driven by activation of the Ras/Erk and Wnt oncogenic pathways, which increase the expression of proliferation genes such as cyclin D and c-Myc (Fig. 1B, red arrows). On the other hand, the TGF-β pathway is known to have potent antiproliferative actions through the activation of cyclin-dependent kinase (CDK) inhibitors such as p14Ink4b and p21WAF1 (reviewed in ref. 6).
As indicated by the green arrows in Fig. 1B, activation of the FGF/EGF/Ras pathway (which activates Erk) and the Wnt pathway (which inhibits GSK3) will result in the accumulation of maximally active Smad4. The consequent increase in the antiproliferative effect of TGF-β signaling would counteract the mitogenic action of Ras and Wnt activation. This protective effect of TGF-β will be lost when the Smad4/DPC4 tumor suppressor is mutated, providing a molecular pathway for the barrier effect of Smad4 during cancer progression. This mechanism explains why the loss of Smad4 has such dramatic effects in mice harboring a truncation of adenomatous polyposis coli (APC),8 as well as in human tumors.6,7 In this regard, Smad4/DPC4 plays such a prominent role in the malignant progression of cancer because the newly identified growth factor-regulated phosphorylations place it at the crossroads of 3 major signaling pathways that balance each other (Fig. 1B).
The discovery that Smad4 activity and stability are regulated directly by MAPK and GSK3 phosphorylations also offers a possible explanation for the decreased stability of Smad4 mutant protein with point mutations that severely inhibit its function. Smad4 is frequently deleted in metastatic tumors, but intragenic point mutations are also quite common.5,9 Several point mutations in the MH1 or MH2 domains have been shown to increase Smad4 degradation by increasing binding of Smad4 to the β-TrCP E3 ubiquitin ligase.10 Our finding that β-TrCP binding to Smad4 is a requirement for GSK3 phosphorylations suggests that in cells harboring point mutations in Smad4 (which possibly cause misfolding) the protein may be hyperphosphorylated by GSK3, generating a phosphodegron that is recognized and bound by the ubiquitin ligase β-TrCP to promote degradation. Ongoing investigations strongly support this model and suggest that some Smad4 mutant proteins commonly found in pancreatic cancer have retained their ability to transduce the TGF-β signal provided that their β-TrCP-mediated degradation is blocked by the addition of GSK3 inhibitors. A potential therapeutic application is that pharmacological GSK3 inhibitors might stabilize Smad4 and restore growth control in tumors harboring such Smad4 mutations.
In conclusion, the finding that Smad4/DPC4 activity and stability are regulated through a set of phosphorylation sites encoded in its primary sequence and by 2 of the most common oncogenic signaling pathways provides a molecular explanation for its barrier role during cancer progression.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by RO1 HD21502–25 and the Howard Hughes Medical Institute, of which E.M.D.R. is an Investigator.
References
- 1. Massague J. TGF β signalling in context. Nat Rev Mol Cell Biol 2012; 13: 616-30; PMID:22992590; http://dx.doi.org/ 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol 2001; 2: 769-76; PMID:11584304; http://dx.doi.org/ 10.1038/35096075 [DOI] [PubMed] [Google Scholar]
- 3. Taelman V, Dobrowolski R, Plouhinec J, Fuentealba L, Vorwald P, Gumper I, Sabatini D, De Robertis E. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010; 143: 1136-48; PMID:21183076; http://dx.doi.org/ 10.1016/j.cell.2010.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demagny H, Araki T, De Robertis EM. The tumor suppressor smad4/DPC4 is regulated by phosphorylations that integrate FGF, Wnt and TGF-β signaling. Cell Rep 2014; 9: 688-700; PMID:25373906; http://dx.doi.org/ 10.1016/j.celrep.2014.09.020 [DOI] [PubMed] [Google Scholar]
- 5. Levy L, Hill C. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev 2006; 17: 41-58; PMID:16310402; http://dx.doi.org/ 10.1016/j.cytogfr.2005.09.009 [DOI] [PubMed] [Google Scholar]
- 6. Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell 2011; 144: 646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 7. Ding Z, Wu C, Chu G, Xiao Y, Ho D, Zhang J, Perry S, Labrot E, Wu X, Lis R, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011; 470: 269-73; PMID:21289624; http://dx.doi.org/ 10.1038/nature09677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin M, Taketo M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998; 92: 645-56; PMID:9506519; http://dx.doi.org/ 10.1016/S0092-8674(00)81132-0 [DOI] [PubMed] [Google Scholar]
- 9. Xu J, Attisano L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor β signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA: 2000; 97: 4820-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang L, Wang N, Tang Y, Cao X, Wan M. Acute myelogenous leukemia-derived SMAD4 mutations target the protein ubiquitin-proteasome degradation. Hum Mutat 2006; 27: 897-905; PMID:16865698; http://dx.doi.org/ 10.1002/humu.20387 [DOI] [PubMed] [Google Scholar]