

Abstract
mTOR inhibition has emerged as a promising strategy for head and neck squamous cell carcinomas (HNSCC) treatment. However, most targeted therapies ultimately develop resistance due to the activation of adaptive survival signaling mechanisms limiting the activity of targeted agents. Thus, co-targeting key adaptive mechanisms may enable more effective cancer cell killing. Here, we performed a synthetic lethality screen using shRNA libraries to identify druggable candidates for combinatorial signal inhibition. We found that the ERK pathway was the most highly represented. Combination of rapamycin with trametinib, a MEK1/2 inhibitor, demonstrated strong synergism in HNSCC-derived cells in vitro and in vivo, including HNSCC cells expressing the HRAS and PIK3CA oncogenes. Interestingly, cleaved caspase-3 was potently induced by the combination therapy in PIK3CA+ cells in vitro and tumor xenografts. Moreover, ectopic expression of PIK3CA mutations into PIK3CA− HNSCC cells sensitized them to the pro-apoptotic activity of the combination therapy. These findings indicate that co-targeting the mTOR/ERK pathways may provide a suitable precision strategy for HNSCC treatment. Moreover, PIK3CA+ HNSCC are particularly prone to undergo apoptosis after mTOR and ERK inhibition, thereby providing a potential biomarker of predictive value for the selection of patients that may benefit from this combination therapy.
Keywords: synthetic lethality screen, shRNA library, rapamycin, trametinib, co-targeting therapy
INTRODUCTION
Recent advances in RNAi technology have enabled synthetic lethal screenings in mammalian cells on a genome-wide scale [1]. Synthetic lethality, first described in yeast genetics, occurs when alteration of a gene results in change of the cellular phenotype only in the presence of another nonlethal genetic alteration. Recently, this approach has been applied to mammalian cells using RNAi screens [2, 3]. For example, RNAi screens in combination with active compounds were used for the identification of sensitizing targets and novel genetic interdependencies in cancer [4, 5]. RNAi screens can be carried out with either siRNA-based transient transfection or shRNA-based stable gene knockdown. Vector-based shRNA libraries have several unique advantages that make them particularly attractive: they can be screened in pools and this significantly reduces the cost of the screen; they afford long-term gene knockdown and thus can reveal slow phenotypic changes in the cell; and they can be readily adapted for in vivo screens in mouse models [1].
Head and neck squamous cell carcinomas (HNSCC) are among the ten cancers most frequently diagnosed each year in the United States, affecting approximately 42,000 new patients and resulting in approximately 8,300 deaths [6]. Mammalian target of rapamycin (mTOR) is at the center of signaling pathways that are critical for the regulation of cellular metabolism, growth, and proliferation [7]. Recent findings indicate that multiple genetic and epigenetic alterations converge on the persistent activation of PI3K/AKT/mTOR signaling in most HNSCC lesions [8-13]. Specifically, activating mutations in the PI3K catalytic subunit α, encoded by the PIK3CA gene, is the most frequent oncogenic event in HNSCC, with 18.1 % of all HNSCC displaying PIK3CA mutations and 21.2 % of cases displaying PIK3CA gene copy number gain. In addition, HNSCC also have multiple genomic alterations, such as PTEN mutations (2.8 %) and gene copy number loss (31.0 %) and activating mutations in RAS (5.9 %) and AKT (2.2 %) genes that result in PI3K/mTOR pathway activation. This cancer driver overreliance may in turn render HNSCC particularly sensitive to PI3K and mTOR inhibitors. Indeed, we and others have demonstrated this pathway dependence in a large series of genetically-defined and chemically-induced preclinical HNSCC experimental models by inhibiting mTOR with rapamycin and its analogs, which inhibit the activity of mTORC1 via binding to FKBP-12 and forming a ternary complex with mTOR [14-18].
The use of rapamycin and rapalogs have validated the concept that the PI3K/AKT/mTOR pathway can be successfully targeted in clinical cancer treatment. In this regard, our recently completed clinical trial using rapamycin in newly diagnosed and previously untreated HNSCC patients has demonstrated promising clinical activity [19] in contrast to most tumor types in which rapalogs often have modest and highly variable responses [18]. However, most targeted agents promote the activation of adaptive cellular responses that ultimately render cancer lesions resistant to their antitumor effect [20]. Thus, the combinatorial use of mTOR inhibitors with other drugs interfering with these resistance mechanisms may represent a promising strategy to improve treatment efficacy. In order to identify new potential targets for combination treatment with mTOR inhibitors, we performed a synthetic lethality screen using a pooled shRNA library with rapamycin in HNSCC cells. We identified a synthetic lethal interaction between ERK pathway inhibition and rapamycin, and validated the synergism of the co-target treatment on the growth inhibition of HNSCC cells in vitro and in vivo. Furthermore, we found that HNSCC cells harboring PIK3CA mutations are particularly susceptible to undergo apoptosis upon mTOR and ERK inhibition, thus providing a new therapeutic option for PIK3CA+ HNSCC patients.
RESULTS
An RNAi screens reveals synthetic lethal interactions with mTOR inhibitors
We utilized a pooled retroviral shRNA library for the synthetic lethal screen [21]. We analyzed the change in relative abundance of each shRNA over time by sequencing of half-hairpin (HH) barcodes derived from the shRNA population to identify those that are antiproliferative and are thus depleted from the population. We compared the lethality signature of the cells in presence of rapamycin and control to identify those shRNAs showing selective depletion in the cells in presence of rapamycin but not in control (Figure 1A). We chose the HNSCC cell line HN12 [16] for the screen, as this HNSCC cell line carries no driver mutations endogenously [9], but, as most HNSCC-derived cells, exhibits elevated mTOR activity and it is sensitive to growth inhibition in response to rapamycin [14]. Target genes were defined as hits when the difference between log2 end/start ratio of rapamycin treated samples and that of control samples was ≤ −2. We identified 117 genes as hits, among which 22 encoded kinases (Figure 1B). This gene set was particularly enriched for kinases involved in ERK pathway signaling, including KSR1 (KSR1), CRAF/RAF1 (RAF1), ERK (MAPK1) and RSK1 (RPS6KA1), which suggests that their depletion by shRNAs is synthetically lethal with rapamycin. We also performed gene ontology (GO) enrichment analysis and found the GO term “MAPK cascade” was significantly related to this gene set (P = 2.7E-8). These findings prompted us to explore the impact of ERK signaling inhibition in HNSCC cells in combination with rapamycin. As an approach, we took advantage that a selective MEK1/2 inhibitor trametinib, which blocks ERK activation, was recently approved for the treatment of unresectable or metastatic melanoma with BRAFV600E or BRAFV600K mutations by the US Food and Drug Administration as a monotherapy and in combination with dabrafenib [22].
Figure 1. RNAi synthetic lethal screen.

A. Schema of shRNA screen. The change in the relative abundance of each shRNA in the library over time is measured using the normalized PD 13/PD 0 ratio of its reads. A log2 PD 13/PD 0 ratio of < 0 indicates the shRNA is depleted in the population over time, and a log2 PD 13/PD 0 ratio of < 0 indicates the shRNA is enriched in the population. B. Hit kinase genes. Targets were filtered by log2 PD 13/PD 0 ratio of < −2 (n = 3). C. Factorial dose matrix combinatorial drug treatment. HN12 cells were incubated for 72 hrs with indicated concentrations of drugs. Numbers on the matrix indicate % Cell Viability (n = 3). D. Computer-simulated Fa-CI curves were created based on the matrix data. The ratios of rapamycin : trametinib were indicated. Synergism (CI < 1), additive effect (CI = 1), or antagonism (CI > 1) for the indicated levels of growth inhibition (Fraction affected) induced by the drug combination. E. mTOR/ERK pathway. HN12 cells were treated with 0.1% DMSO, 20nM rapamycin, 20nM trametinib or the combination for 24hrs.
In order to evaluate the effect of drug-drug interaction on cell viability, we performed a factorial dose matrix combinatorial drug treatment with rapamycin and trametinib. HN12 cells were incubated with these drugs in a 6 × 8 dose-response matrix and cell viability was measured after 72 hrs of treatment (Figure 1C). We also investigated whether the combination effect displayed a synergistic activity. For this purpose, the fraction affected (Fa) combination index (CI) plot (Fa-CI plot) curves were simulated using CompuSyn software [23]. CI at 0.5 of Fa was 0.14 and 0.10 for 1:4 and 1:16 constant ratio of rapamycin to trametinib respectively (Figure 1D). These data indicated a strong synergism of the combination therapy in HN12 cells. We next evaluated the effect of rapamycin and trametinib on the ERK and PI3K-mTOR signaling pathway in these HNSCC cells. For this purpose, cells were seeded in 6-well plates and harvested after 24h treatment, and samples were used for western blotting analysis. As expected, rapamycin decreased phospho-S6 (pS6) and phospho-4E-BP1 (p4E-BP1) (Figure 1E). In contrast, phospho-AKT (pAKT) and phospho-ERK (pERK) were increased (Figure 1E). Instead, trametinib clearly decreased pERK, consistent with its MEK blocking activity, and of interest, trametinib also partially decreased pS6 (Figure 1E).
Synergistic effects of the combination of rapamycin and trametinib on HNSCC harboring HRAS and PIK3CA mutations
HNSCC lesions often harbor activating mutations in PIK3CA (18.1 %), encoding the catalytic PI3K-α subunit and less frequent oncogenic mutants of the HRAS (5.6 %) or KRAS (0.2 %) genes [8, 12], collectively referred herein as RAS. To explore whether the combination of rapamycin and trametinib also displays synergistic effect in HNSCC tumors harboring activating mutations of RAS and PIK3CA, we evaluated the impact of this drug combination in HNSCC cells harboring endogenous oncogenic mutations. We used UM-SCC-17B, which has a HRAS Q61L mutant and Detroit 562 cells exhibiting the PIK3CA H1047R mutation. The combination of rapamycin and trametinib demonstrated strong synergism in growth inhibition of these cells (Figure 2A, Supplementary Figure S1A). We next evaluated the effect of the combination treatment in the downstream signaling pathway of RAS and PI3K in these cell lines. Rapamycin decreased pS6 and p4E-BP1, but in contrast, pAKT and pERK were both increased (Figure 2B). Trametinib treatment clearly decreased pERK, and it also enhanced the rapamycin-mediated decrease of pS6 and p4E-BP1 (Figure 2B). We also evaluated whether trametinib, rapamycin, or their combination can promote apoptosis as judged by the accumulation of cleaved caspase-3. Remarkably, we found that whereas these drugs alone have limited effect, the combination treatment induced a significant increase in apoptosis in PIK3CA mutant HNSCC cell line, Detroit 562, but not in the HRAS mutant HNSCC cells UM-SCC-17B (Figure 2C).
Figure 2. Synergistic effects of the combination of rapamycin and trametinib on HNSCC-derived cells harboring HRAS or PIK3CA mutations.

A. Computer-simulated Fa-CI curves were created based on the factorial dose matrix combinatorial drug treatment. The ratios of rapamycin : trametinib were indicated. Synergism (CI < 1), additive effect (CI = 1), or antagonism (CI > 1) for the indicated levels of growth inhibition (Fraction affected) induced by the drug combination. B. mTOR/ERK signaling pathway. Cells were treated with 0.1% DMSO, 20nM rapamycin, 20nM trametinib or the combination for 24hrs. C. Immunoblot analysis for cleaved-PARP and cleaved caspase-3. Cells were treated with 0.1% DMSO, 20nM rapamycin, 20nM trametinib or the combination for 24hrs.
Antitumoral activity of the combination therapy with rapamycin and trametinib against HNSCC harboring HRAS or PIK3CA mutations
We next evaluated the antitumoral activity of the ERK-mTOR targeting combination therapy in vivo, using UM-SCC-17B and Detroit 562 HNSCC xenografts, which harbor endogenous activating HRAS and PIK3CA mutations, respectively. The combination therapy with rapamycin and trametinib was more effective than each of the single agents individually (Figure 3A). The body weight after 4 weeks treatment was 22.7±2.1 g in the combination treatment group and 23.7±0.7 g in vehicle control group. The antitumoral activity was reflected by a decreased immunohistochemical detection of nuclear Ki67, which was used for the evaluation of cell proliferation. The MEK-mTOR combination therapy showed significant reduction of HNSCC cells expressing nuclear Ki67 in both xenograft models (Figure 3B). Immunohistochemistry analysis of treated tumors revealed that trametinib enhanced the rapamycin-mediated inhibition of S6 and 4E-BP1 in vivo, and blunted the rapamycin-induced ERK activation in these HNSCC cells (Figure 4A, 4B). Consistent with the activation of ERK, rapamycin also induced elevated phospho-MEK1/2 (pMEK1/2) levels. pMEK1/2 was increased by trametinib, likely as a feedback mechanisms stimulating the activation of MEK upstream kinases upon MEK inhibition by its direct blocker. AKT was activated by rapamycin in UM-SCC-17B but not in Detroit 562 in vivo, suggesting that activation of the ERK pathway may represent a more general secondary effect of rapamycin than pAKT increase (Figure 4A). In addition, a clear distinction between these HNSCC cells was that cleaved caspase-3 was significantly increased by the combination therapy in Detroit 562 (PIK3CA mutant) but not in UM-SCC-17B (HRAS mutant) (Figure 4C).
Figure 3. Antitumoral activity of the mTOR ERKcombination therapy with rapamycin and trametinib against HNSCC harboring HRAS or PIK3CA mutations.

A. Antitumor efficacy of rapamycin, trametinib, and combination. Athymic nude mice were transplanted with HNSCC cells. Treatment was initiated when the tumor volume reached approximately 200 mm3. The tumor growth curves (left), tumor weights at the end of the single agent and combined treatments (middle) and pictures of representative tumors (right) are displayed. Data points represent mean values ± SE (n = 10 per each group). B. Representative tumor tissue sections (top) and quantification (bottom) stained for Ki67 (n = 6 per each group). Scale bars represent 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 4. Effects of the combination of rapamycin and trametinib on mTOR/ERK signaling and apoptosis in HNSCC harboring HRAS or PIK3CA mutations.
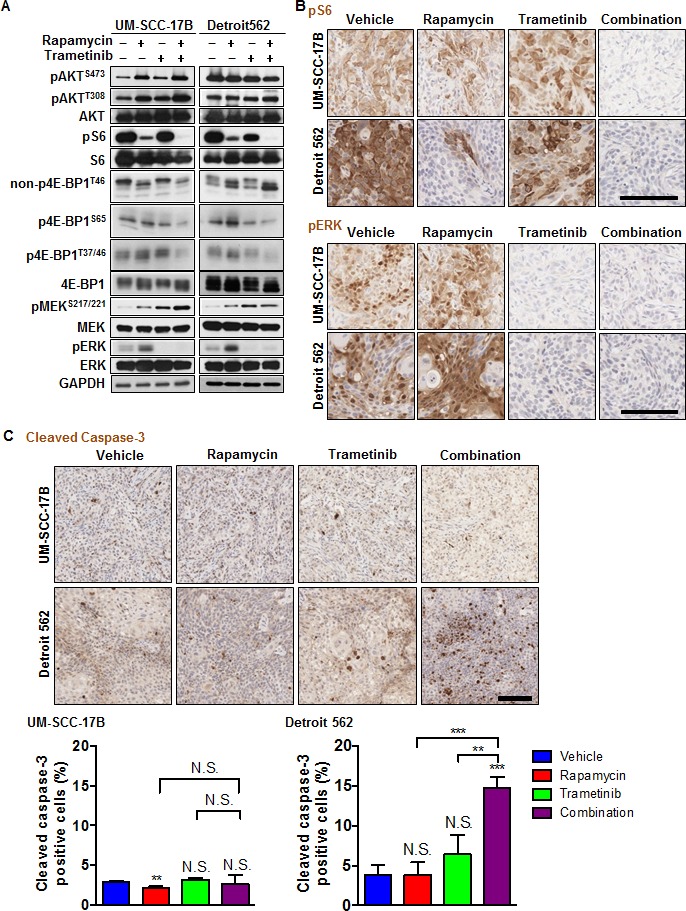
A. mTOR/ERK signaling pathway. Cells were transplanted into athymic mice and treated with vehicle, rapamycin, trametinib or combination for four days. Tumor lysates were analyzed with immunoblot analysis. B. Representative tumor tissue sections stained for pS6 and pERK. Scale bars represent 100 μm. C. Representative tumor tissue sections (top) and quantification (bottom) stained for cleaved-caspase 3 (n = 3 per each group). Scale bars represent 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
mTOR and MEK inhibition display a synergistic growth inhibitory activity in HNSCC cells genetically engineered to express activating KRAS and PIK3CA mutations
Our observations suggested that mTOR and MEK co-targeting exerts a synergistic antiproliferative effect in HNSCC cells in vitro and in vivo, and a selective pro-apoptotic impact on PIK3CA expressing HNSCC cells. However, cancer cells display an intrinsic heterogeneity, hence it is unclear if trametinib and rapamycin promote apoptosis in Detroit 562 because these cells express mutant PIK3CA or if this response is due to cell-dependent variations. As an approach to explore these possibilities, we next investigated whether a significant synergism was displayed in isogenic HNSCC cells, CAL27 cells, which were genetically engineered to express RAS and PIK3CA mutants [17]. Strong synergism was demonstrated between trametinib and rapamycin in wild type CAL27 cells and in both KRAS and PIK3CA expressing CAL27 cells, supporting that the drug combination is effective in HNSCC cells displaying multiple genetic alterations (Figure 5A, Supplementary Figure S1B). In this biological relevant system, PI3KCA expressing CAL27 cells showed significantly stronger synergism than CAL27 WT and CAL27 KRAS cells. Indeed, CI at ED50 was 0.09, 0.06 and 0.01 for CAL27 WT, CAL27 KRAS and CAL27 PIK3CA cells, respectively (Figure 5A).
Figure 5. Synergism of rapamycin and trametinib in genetically engineered HNSCC cells to express activating KRAS or PIK3CA mutations.

A. Computer-simulated Fa-CI curves were created based on the factorial dose matrix combinatorial drug treatment. The ratios of rapamycin : trametinib were 1:16. Synergism (CI < 1), additive effect (CI = 1), or antagonism (CI > 1) for the indicated levels of growth inhibition (Fraction affected) induced by the drug combination (left). CI values at Fa = 0.5 was used to calculate the mean between experiments (n = 3). *P < 0.05, **P < 0.01. B. mTOR/ERK signaling pathway. Cells were treated with 0.1% DMSO, 20nM rapamycin, 20nM trametinib or the combination for 0.5h or 24hrs. C. Immunoblot analysis for cleaved-caspase 3. Cells were treated with 0.1% DMSO, 20nM rapamycin, 20nM trametinib or the combination for 24hrs.
We next evaluated the effect of rapamycin and trametinib in the downstream signaling pathway elicited by RAS and PI3K in these cells. As for other HNSCC cells, rapamycin decreased pS6 and p4E-BP1 in these cell lines. In contrast, pAKT and pERK were increased (Figure 5B). Trametinib clearly decreased pERK and enhanced the rapamycin-mediated reduction of pS6 and p4E-BP1 (Figure 5B). We also evaluated apoptosis by cleaved caspase-3 (Figure 5C). Rapamycin increased caspase-3 cleavage in CAL27 PIK3CA cells but failed to stimulate apoptosis in the CAL27 WT cell line. Trametinib also promoted a limited increase in cleaved caspase-3 in CAL27 KRAS, and this effect was much more remarkable in CAL27 PIK3CA cells (Figure 5C).
Antitumoral activity of the rapamycin and trametinib combination therapy in genetically engineered HNSCC cells expressing activating RAS or PIK3CA mutations
We next investigated whether the combination of rapamycin and trametinib was effective in the isogenic cell line panel CAL27 WT, CAL27 HRAS and CAL27 PIK3CA xenograft models. A more significant antitumoral effect was observed in the groups treated with the combination therapy than in the groups treated with single agents in each of these xenograft models (Figure 6A). This was reflected by decreased nuclear Ki67, a biomarker for cell proliferation, in the combination therapy compared to single agents (Figure 6B, Supplementary Figure S2A). To study the biochemical and biological effects of the drug combination in vivo in this isogenic HNSCC cell panel, we performed immunohistochemistry for pS6, pERK (Figure 7A) and cleaved caspase-3 (Figure 7B). Rapamycin mediated inhibition of pS6 was enhanced by the combination with trametinib (Figure 7A). ERK was activated by rapamycin in these cells, and strongly inhibited by the therapy with trametinib (Figure 7A). Cleaved caspase-3 was significantly induced in the tumors treated with the combination therapy in CAL27 PIK3CA (Figure 7B). However, only minor pro-apoptotic effects were observed in CAL27 WT and CAL27 KRAS (Figure 7B, Supplementary Figure S2B). These finding suggest that while co-targeting MEK and mTOR is effective for tumor reduction in all HNSCC models tested, PIK3CA expression specifically sensitizes HNSCC cells to the pro-apoptotic activity of MEK and mTOR combined targeted treatment, thereby displaying increased response.
Figure 6. Antitumoral activity of the rapamycin and trametinib combination therapy against genetically engineered HNSCC cells expressing KRAS and PIK3CA oncogenes.

A. Antitumor efficacy of rapamycin, trametinib, and combination. Athymic nude mice were transplanted with HNSCC cells. Treatment was initiated when the tumor volume reached approximately 200 mm3. The tumor growth curves (left), tumor weights at the end of the single agent and combined treatments (middle) and representative histological sections for each treatment group. Scale bars represent 5 mm (right) are displayed. Data points represent mean values ± SE (n = 10 per each group). B. Representative tumor tissue sections (top) and quantification (bottom) stained for Ki67 (n = 6 per each group). Scale bars represent 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 7. Effects of the combination of rapamycin and trametinib on mTOR/ERK signaling and apoptosis in genetically engineered HNSCC cells KRAS and PIK3CA mutations.
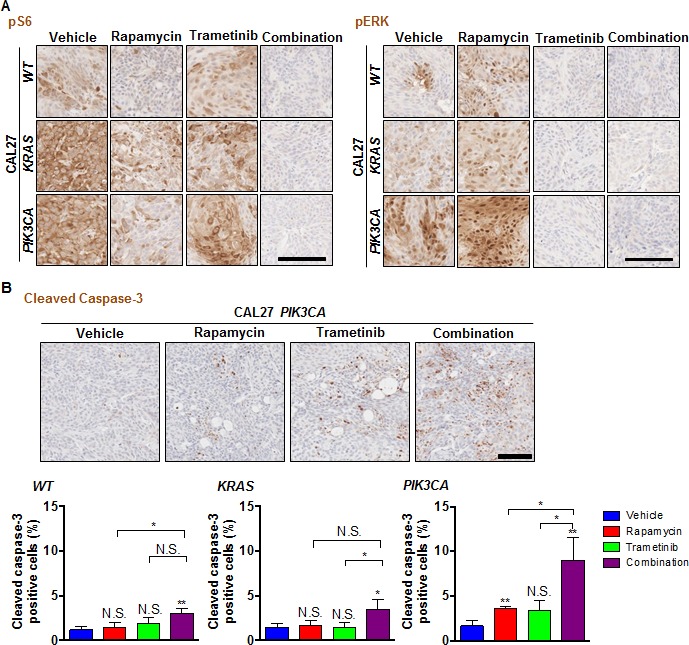
A. Representative tumor tissue sections stained for pS6 and pERK. Scale bars represent 100 μm. B. Representative tumor tissue sections (top) and quantification (bottom) stained for cleaved-caspase 3 (n = 3 per each group). Scale bars represent 100 μm. *P < 0.05, **P < 0.01.
DISCUSSION
Our in vitro data demonstrated strong synergism of the combination treatment with rapamycin and trametinib against HNSCC -derived cells exhibiting quite distinct genomic alterations, including cells carrying endogenous and genetically engineered RAS or PIK3CA mutations. Furthermore, the co-targeting therapy demonstrated significant effect on tumor growth in all HNSCC xenograft models tested. Intriguingly, CAL27 engineered to express PIK3CA mutations displayed the strongest synergism in vitro. Furthermore, the combination treatment significantly induced cleaved-caspase 3 in HNSCC cell lines carrying PIK3CA mutation endogenously and when ectopically expressed. Thus, in the presence of PIK3CA mutations, adding MEK inhibitors to mTOR blockade causes a proapoptotic switch from cytostatic to cytotoxic, which has important beneficial therapeutic implications.
The mechanism of this remarkable genotype-specific drug interaction remains unclear. For example, the ERK pathway has profound effects on the regulation of cell survival pathways by the post-translational phosphorylation of pro-apoptotic and apoptotic regulatory molecules, including BAD, BIM, Mcl-1, caspase 9 and Bcl-2 [24]. On the other hand, mTOR also possesses both pro-apoptotic and anti-apoptotic effects [25]. Activated S6K is capable of binding to mitochondrial membrane and phosphorylating the pro-apoptotic molecule BAD, rendering BAD inactive and promoting cell survival [26]. In line with these observations, mTOR inhibition can promote apoptosis in several cancer models [16, 27]. Conversely, several studies demonstrated that mTOR inhibition attenuates cytotoxic agent-induced apoptosis in malignant cells [28, 29]. The mTOR function on apoptosis appears to be dictated by cell type and its activation state as well as by the mutational status of its downstream targets including well known apoptosis-regulatory proteins such p53, BAD and Bcl-2 [30]. Thus, the interplay between mTOR and ERK inhibition may have pro-apoptotic functions in restricted genetic contexts. In line with this possibility, recently, Hata et al. reported that the concomitant use of MEK and PI3K inhibitor lead to upregulation of PUMA and BIM, both of which were necessary for the induction of an apoptotic response in KRAS-mutant in another cancer type, non-small cell lung cancer (NSCLC) [31, 32]. Certainly, the underlying reasons in HNSCC that an apoptotic response is only elicited in PIK3CA mutant cells is clearly clinically relevant, and its precise underlying mechanism warrants further investigation.
In the current study we identified 22 kinases whose suppression is synthetic lethal with rapamycin for therapy against HNSCC. These include KSR1 (KSR1), CRAF/RAF1 (RAF1), ERK (MAPK1) and RSK1 (RPS6KA1), which are involved in the ERK signaling pathway, suggesting that interfering with ERK activation renders HNSCC cells more sensitive to mTOR inhibition. Indeed, we show here that co-targeting MEK and mTOR using FDA approved therapeutic agents, trametinib and rapamycin, respectively, exerts a synergistic anti-proliferative and pro-apoptotic effect in all HNSCC cell lines tested irrespective of their quite distinct mutational genomic landscape [9]. Furthermore, we found that the combined drug therapy was much more effective in promoting tumor regression that any of the single agents alone, supporting that synthetic gene lethality screens can be exploited for the development of new therapeutic options for HNSCC treatment.
In addition to the ERK pathway, we also identified p38 MAPK (MAPK14) and MAP3K2 (MAP3K2), which are involved in other MAPK pathways as co-targeting candidates. Recently, we reported that p38 MAPK functions as a positive regulator of HNSCC in the context of the tumor microenvironment, controlling cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis [33]. We also identified kinases involved in PI3K/AKT pathway. Among these were SGK3 (SGK3), ARK5/NUAK1 (NUAK1), and NDR2 (STK38L). For example, activation of SGK3 downstream of PIK3CA and INPP4B is required for 3D cell proliferation, invasive migration, and tumorigenesis in vivo [34]. Overexpression of ARK5 confers tolerance to glucose starvation, which is a stress that leads to a decrease in ATP and an increase in AMP [35]. Activated AKT stimulates cell invasion by phosphorylating ARK5 at Ser600 [35]. ARK5 has been also reported to act as a tumor invasion-associated factor through NDR2 during IGF-1 signaling [36]. We also identified FAK (PTK2) and HIPK2 (HIPK2) in our synthetic lethality screens. FAK promotes cell survival through kinase-dependent and kinase-independent mechanisms [37, 38]. Overall, these results suggest that several MAPK and PI3K/AKT pathway components and FAK, among others, may participate in adaptive response mechanisms promoting resistance to rapamycin in HNSCC cells. Hence, their knock down or pharmacological inhibition with small molecule inhibitors can be also considered for the future development of more effective combination therapies for HNSCC.
Overall, our unbiased genetic screen revealed that co-targeting mTOR and ERK using FDA approved agents results in a remarkable synergistic interaction in HNSCC preclinical models. While the inhibition of the PI3K/AKT/mTOR and ERK pathways has been investigated in other malignancies, the in vivo impact of this co-targeting strategy for HNSCC has not been well validated. Indeed, in all HNSCC tumors tested, the combination of tramatinib and rapamycin caused a rapid tumor collapse, and was much more active than any of these agents alone. Furthermore, we now show that the concomitant inhibition of mTOR and MEK promotes the apoptotic demise of HNSCC cells specifically in tumors expressing the PIK3CA oncogene, the most frequently driver mutation in this cancer type [8, 12]. Thus, these findings will have significant clinical implications, as they define a new mechanism-based precision therapeutic approach for the treatment of HNSCC patients, and define PIK3CA as a suitable biomarker of heightened beneficial therapeutic response to the combined treatment.
MATERIALS AND METHODS
Cell lines, tissue culture and antibodies
Human head and neck cancer cell lines Cal27, Detroit 562, were from American Type Culture Collection; UM-SCC-17B cell line was from Thomas Carey, University of Michigan. Cells were cultured in Dulbecco's modified Eagle's medium with 10 % fetal bovine serum supplemented with antibiotics, 5 % CO2 at 37°C. All cell lines underwent DNA authentication by multiplex STR profiling (Genetica DNA Laboratories, Inc. Burlington, NC) prior to the described experiments to ensure consistency in cell identity. Antibodies against AKT, pAKTS473, pAKTT308, ERK1/2, pERK1/2, S6, pS6, MEK1/2, pMEK1/2, 4E-BP1, non-p4E-BP1T46, p4E-BP1T37/46, p4E-BP1S65, cleaved caspase-3, cleaved PARP, α-Tubulin-HRP, GAPDH were purchased from Cell Signaling Technology (Beverly, MA). Mouse anti-Ki67 antibody was purchased from DAKO (Carpinteria, CA).
shRNA screening
shRNA screening was performed as described [21]. HN12 cells were infected with pools of retroviral shRNA at a representation of ∼1,000 and a multiplicity of infection (MOI) of ∼1. At day 3 post infection cells were selected with puromycin for an additional 3 days (1 μg/ml) to remove the minority of uninfected cells. After that, cells where propagated in culture for 3 days and then an initial population-doubling 0 (PD 0) sample was taken. The rest of the population was divided in 6 groups, and 3 groups where kept as a control and 3 where treated with rapamycin (100 nM). Cells where propagated in the presence or not of drug for an additional 12 doublings before the final, PD 13 sample was taken. For each passage a minimal representation of 1000 was maintained. The shRNA library contained 9,149 retroviral shRNAs targeting human kinases, phosphatases, genes involved in protein ubiquination, and genes implicated in cancer. The libraries where expressed using the retroviral vector MSCV-PM. For PD 0 and PD 13 samples, shRNA HH barcode was PCR-recovered from genomic samples and samples where sequenced to calculate abundance of the different shRNA probes.
The change in the relative abundance of each shRNA in the library over time is measured using the normalized PD 13/PD 0 ratio of its reads. A log2 PD 13/PD 0 ratio of < 0 indicates the shRNA is depleted in the population over time, and a log2 PD 13/PD 0 ratio of < 0 indicates the shRNA is enriched in the population. To identify shRNAs that are synthetically lethal with rapamycin, the mean log2 PD 13/PD 0 ratios of the rapamycin treated cell triplicates were compared to that of the control triplicates to derive the log2 ratio difference. A p-value of the difference between the two triplicates was calculated using the t-test. Targets were filtered by the presence of at least 2 different shRNAs for the same gene and a P < 0.05. GO enrichment analysis was performed with ToppGene Suite software[39].
Cell growth assays and drug combination analysis
Alamar Blue Cell Viability Reagent was purchased from Life Technologies. (Grand Island, NY) Cells were cultured in 96-well-plate and treated with drugs for 72 hours. The manufacturer's instructions were followed to complete the assay. Drug synergy was determined by the combination index methods using CompuSyn software [23]. CI values less than 1, equal to 1, and greater than 1 represent synergism, additivity, and antagonism, respectively.
Immunoblot analysis
The cells and the tissues were lysed in RIPA buffer containing Halt protease and phosphatase inhibitor cocktail (Thermo Scientific, Rockford, IL). Protein concentration was measured by Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Equal amounts of total proteins were subjected to SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes. Membranes were blocked with 5% nonfat dry milk in T-TBS buffer (50 mM Tris/HCl, pH 7.5, 0.15 M NaCl, 0.1% [v/v] Tween-20) for 2 h, and then incubated with primary antibodies in blocking buffer at 4°C overnight. Detection was conducted by incubating the membranes with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Southern Biotech, Birmingham, AL) was used at a dilution of 1:50,000 in 5 % milk-T-TBS buffer, at room temperature for 1 h, and visualized with Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA).
Tissue analysis
All samples were fixed in zinc formalin (Z-Fix, Anatech) and embedded in paraffin; 5 μm sections were stained with Hematoxylin-Eosin for diagnostic purposes. For immunohistochemistry (IHC) studies the sections were deparaffinized, hydrated through graded ethanols, and the endogenous peroxidase inhibited with 3 % H2O2 in 70 % ethanol. The slides were extensively washed with distilled water and antigen retrieval was performed by high temperature treatment with 10 mM citric acid in a microwave. After washing with water and PBS, the slides were successively incubated with the primary and secondary antibodies, and the ABC reagent (Vector Laboratories, Burlingame, CA). The reaction was developed with 3-3′-diamonobenzifdine under microscopic control. A Mouse on Mouse (M.O.M.) Basic Kit (Vector Laboratories, Burlingame, CA) was used in Ki67 staining to inhibit binding of secondary antibodies to mouse tissue.
In vivo mouse experiments and analysis
All the mice studies were carried out according to National Institutes of Health (NIH) approved protocols (ASP #13-695) in compliance with the NIH Guide for the Care and Use of Laboratory Mice. To establish tumor xenografts, cells were transplanted into the flanks of athymic nude mice (female, four to six weeks old) (Harlan Laboratories, Inc., Indianapolis, IN), and when the tumor volume reached approximately 200 mm3, the mice were randomized into groups and treated by intraperitoneal injection (ip) with trametinib (1 mg/kg/day) and rapamycin (5 mg/kg/day), or control diluent (n = 10 per each group). Tumor volume was calculated by using the formula length × width × width/2. The mice were euthanized at the indicated time points and tumors isolated for histologic and immunohistochemical evaluation.
Data and statistical analysis
Data were analyzed using GraphPad Prism version 6 (GraphPad Software, San Diego, CA). Comparisons between experimental groups were made using unpaired t test. P < 0.05 was considered to be statistically significant.
SUPPLEMENTARY MATERIAL FIGURES
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Hu G, Luo J. A primer on using pooled shRNA libraries for functional genomic screens. Acta biochimica et biophysica Sinica. 2012;44:103–112. doi: 10.1093/abbs/gmr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simons A, Dafni N, Dotan I, Oron Y, Canaani D. Establishment of a chemical synthetic lethality screen in cultured human cells. Genome research. 2001;11:266–273. doi: 10.1101/gr.154201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simons AH, Dafni N, Dotan I, Oron Y, Canaani D. Genetic synthetic lethality screen at the single gene level in cultured human cells. Nucleic acids research. 2001;29:E100. doi: 10.1093/nar/29.20.e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernards R, Brummelkamp TR, Beijersbergen RL. shRNA libraries and their use in cancer genetics. Nature methods. 2006;3:701–706. doi: 10.1038/nmeth921. [DOI] [PubMed] [Google Scholar]
- 5.Seyhan AA, Varadarajan U, Choe S, Liu Y, McGraw J, Woods M, Murray S, Eckert A, Liu W, Ryan TE. A genome-wide RNAi screen identifies novel targets of neratinib sensitivity leading to neratinib and paclitaxel combination drug treatments. Molecular bioSystems. 2011;7:1974–1989. doi: 10.1039/c0mb00294a. [DOI] [PubMed] [Google Scholar]
- 6.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 7.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nature reviews Molecular cell biology. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 8.Iglesias-Bartolome R, Martin D, Gutkind JS. Exploiting the head and neck cancer oncogenome: widespread PI3K-mTOR pathway alterations and novel molecular targets. Cancer discovery. 2013;3:722–725. doi: 10.1158/2159-8290.CD-13-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin D, Abba MC, Molinolo AA, Vitale-Cross L, Wang Z, Zaida M, Delic NC, Samuels Y, Lyons JG, Gutkind JS. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget. 2014;5:8906–8923. doi: 10.18632/oncotarget.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molinolo AA, Marsh C, El Dinali M, Gangane N, Jennison K, Hewitt S, Patel V, Seiwert TY, Gutkind JS. mTOR as a molecular target in HPV-associated oral and cervical squamous carcinomas. Clinical cancer research. 2012;18:2558–2568. doi: 10.1158/1078-0432.CCR-11-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral oncology. 2009;45:324–334. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas N Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hedberg ML, Goh G, Chiosea SI, Bauman JE, Freilino ML, Zeng Y, Wang L, Diergaarde BB, Gooding WE, Lui VW, Herbst RS, Lifton RP, Grandis JR. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. The Journal of clinical investigation. 2015 [Google Scholar]
- 14.Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, Molinolo AA, Gutkind JS. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer research. 2005;65:9953–9961. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 15.Amornphimoltham P, Leelahavanichkul K, Molinolo A, Patel V, Gutkind JS. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clinical cancer research. 2008;14:8094–8101. doi: 10.1158/1078-0432.CCR-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amornphimoltham P, Patel V, Leelahavanichkul K, Abraham RT, Gutkind JS. A retroinhibition approach reveals a tumor cell-autonomous response to rapamycin in head and neck cancer. Cancer research. 2008;68:1144–1153. doi: 10.1158/0008-5472.CAN-07-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Martin D, Molinolo AA, Patel V, Iglesias-Bartolome R, Degese MS, Vitale-Cross L, Chen Q, Gutkind JS. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. Journal of the National Cancer Institute. 2014:106. doi: 10.1093/jnci/dju215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng LH, Zheng XF. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta pharmacologica Sinica. 2015;36:1163–9. doi: 10.1038/aps.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirai K, Day TA, Szabo E, Van Waes C, O'Brien PE, Matheus MG, Godwin K, Sood AJ, Vick JA, Martin D, Vitale-Cross L, Molinolo A, Gutkind JS. A pilot, single arm, prospective trial using neoadjuvant rapamycin prior to definitive therapy in head and neck squamous cell carcinoma. Journal of Clinical Oncology. 2015:33. [Google Scholar]
- 20.Rebecca VW, Smalley KS. Change or die: targeting adaptive signaling to kinase inhibition in cancer cells. Biochemical pharmacology. 2014;91:417–425. doi: 10.1016/j.bcp.2014.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandal R, Becker S, Strebhardt K. Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: an emerging threat to anticancer therapy. Oncogene. 2015 doi: 10.1038/onc.2015.329. [DOI] [PubMed] [Google Scholar]
- 23.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 24.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochimica et biophysica acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen SA, Walker D, Gillespie MB, Gutkind JS, Day TA. mTOR inhibitors and its role in the treatment of head and neck squamous cell carcinoma. Current treatment options in oncology. 2012;13:71–81. doi: 10.1007/s11864-011-0180-2. [DOI] [PubMed] [Google Scholar]
- 26.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephan S, Datta K, Wang E, Li J, Brekken RA, Parangi S, Thorpe PE, Mukhopadhyay D. Effect of rapamycin alone and in combination with antiangiogenesis therapy in an orthotopic model of human pancreatic cancer. Clinical cancer research. 2004;10:6993–7000. doi: 10.1158/1078-0432.CCR-04-0808. [DOI] [PubMed] [Google Scholar]
- 28.Calastretti A, Bevilacqua A, Ceriani C, Vigano S, Zancai P, Capaccioli S, Nicolin A. Damaged microtubules can inactivate BCL-2 by means of the mTOR kinase. Oncogene. 2001;20:6172–6180. doi: 10.1038/sj.onc.1204751. [DOI] [PubMed] [Google Scholar]
- 29.Markova B, Hahnel PS, Kasper S, Herbertz S, Schuler M, Breitenbuecher F. Pharmacologic inhibition of mTOR antagonizes the cytotoxic activity of pemetrexed in non-small cell lung cancer. Journal of cancer research and clinical oncology. 2012;138:545–554. doi: 10.1007/s00432-011-1123-9. [DOI] [PubMed] [Google Scholar]
- 30.Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell death and differentiation. 2002;9:99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- 31.Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA, Walton Z, Sarosiek KA, Letai A, Heist RS, Mino-Kenudson M, Wong KK, Engelman JA. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer research. 2014;74:3146–3156. doi: 10.1158/0008-5472.CAN-13-3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hata AN, Engelman JA, Faber AC. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer discovery. 2015;5:475–487. doi: 10.1158/2159-8290.CD-15-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leelahavanichkul K, Amornphimoltham P, Molinolo AA, Basile JR, Koontongkaew S, Gutkind JS. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Molecular oncology. 2014;8:105–118. doi: 10.1016/j.molonc.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Molecular cell. 2014;56:595–607. doi: 10.1016/j.molcel.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun X, Gao L, Chien HY, Li WC, Zhao J. The regulation and function of the NUAK family. Journal of molecular endocrinology. 2013;51:R15–22. doi: 10.1530/JME-13-0063. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki A, Ogura T, Esumi H. NDR2 acts as the upstream kinase of ARK5 during insulin-like growth factor-1 signaling. The Journal of biological chemistry. 2006;281:13915–13921. doi: 10.1074/jbc.M511354200. [DOI] [PubMed] [Google Scholar]
- 37.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nature reviews Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. The Journal of clinical investigation. 2009;119:252–266. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic acids research. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.