

Abstract
The important human pathogen Staphylococcus aureus is able to satisfy its nutrient iron requirement by acquiring heme from host hemoglobin in the context of infection. However, heme acquisition exposes S. aureus to heme toxicity. In order to detect the presence of toxic levels of exogenous heme, S. aureus is able to sense heme through the heme sensing system (HssRS) two-component system. Upon sensing heme, HssRS directly regulates the expression of the heme-regulated ABC transporter HrtAB, which alleviates heme toxicity. Importantly, the inability to sense or respond to heme alters the virulence of S. aureus, highlighting the importance of heme sensing and detoxification to staphylococcal pathogenesis. Furthermore, potential orthologues of the Hss and Hrt systems are found in many species of Gram-positive bacteria, a possible indication that heme stress is a challenge faced by bacteria whose habitats include host tissues rich in heme.
Staphylococcus aureus Pathogenesis
Staphylococcus aureus is a Gram-positive bacterial pathogen that is responsible for a tremendous amount of morbidity and mortality [1]. S. aureus is the most frequent causative agent of hospital-acquired infections and community-acquired infections that necessitate admission to emergency rooms in the USA [2]. Antibiotic-resistant strains of S. aureus such as community-acquired methicillin-resistant S. aureus (CA-MRSA) are increasingly prevalent, even among otherwise healthy individuals [3, 4]. This point is underscored by the fact that MRSA infections are now responsible for more US deaths annually than the human immunodeficiency virus [3, 5]. Taken together, these facts have galvanized efforts aimed at identifying novel therapeutic targets against S. aureus. One area that has received significant attention in this regard is the identification of factors that allow S. aureus to adapt to and survive within the hostile host environment.
A commensal of the skin and anterior nares, S. aureus enters tissues of the host when epithelial barriers become compromised [6–8]. Following tissue invasion, S. aureus is able to cause infection of virtually any anatomical site leading to diseases ranging from skin and soft tissue infections to endocarditis, osteomyelitis, pneumonia, meningitis, and septicemia. Regardless of the site of infection, S. aureus invariably encounters host blood during infection. Blood is the bearer of cells of the immune system that are important for combating staphylococcal infection, as well as the carrier of nutrients that invasive bacteria must acquire in order to colonize and proliferate within host tissues [9–11].
Staphylococcal Heme Metabolism
One nutrient that S. aureus must acquire in the context of infection is iron. Iron is required for a number of cellular processes including nucleotide biosynthesis, aerobic respiration, and resistance to reactive oxygen species [12, 13]. Within host tissues, free iron is actively maintained at concentrations that are too low to support the growth of S. aureus and most other bacterial pathogens [12]. Nonetheless, host tissues are rich in other forms of iron, such as iron in the context of the porphyrin heme. Heme is the cofactor for hemoglobin within erythrocytes and myoglobin within myocytes and therefore is highly abundant within the host [12, 14]. In order to satisfy its nutrient iron requirement during infection, S. aureus expresses a set of cell wall-anchored, membrane-associated, and cytoplasmic proteins that are devoted to the acquisition of iron from heme associated with hemoglobin [9, 10]. Designated the iron-regulated surface determinants (Isd) and the heme transport system (Hts), these factors bind hemoglobin, remove the cofactor heme, traffic heme across the cell wall, import heme into the cytoplasm, and degrade heme to release free iron to satisfy nutrient needs [9, 10, 15–17]. Mutation of Isd or Hts family members attenuates the virulence of S. aureus, highlighting the importance of heme acquisition to staphylococcal pathogenesis [17, 18].
The Adaptive Response of S. aureus to Heme
Conceptually, heme acquisition on the part of a bacterial pathogen raises an interesting paradox. Although a number of bacterial pathogens express systems dedicated to the acquisition of heme during infection, heme is a toxic molecule that is capable of generating reactive oxygen species and damaging cells. Therefore, biological systems that acquire, synthesize, or associate with heme must also possess systems devoted to the avoidance of heme toxicity [10, 19–22]. Given the significant association of S. aureus with blood during infection and the efficiency of the staphylococcal heme uptake machinery, it is likely that S. aureus expresses systems that protect this bacterium from the toxic side effects of heme acquisition during infection.
An initial indication that S. aureus may express a heme detoxification system(s) came from experiments designed to test whether S. aureus is sensitive to the toxic effects of heme and whether the bacterium is able to respond accordingly [23]. These experiments revealed two important observations. First, S. aureus is indeed sensitive to heme toxicity. When S. aureus is subcultured into medium containing a high heme concentration, no bacterial proliferation occurs as measured by growth curve analysis due to a bactericidal activity that heme exerts on S. aureus [23]. Second, S. aureus is able to sense the presence of exogenous heme and adapt to growth in its presence. This can be demonstrated by pre-exposing S. aureus to a sub-toxic concentration of heme, then subculturing the bacteria into medium containing an otherwise toxic heme concentration and monitoring bacterial proliferation. When staphylococci are pre-exposed to heme, appreciable growth occurs and cells are more resistant to the bactericidal activity of heme [23]. These results suggest that S. aureus is capable of sensing and adapting to the presence of exogenous heme, presumably through a coordinated change in the expression of a heme detoxification system.
The Heme Regulated Transporter (HrtAB)
To test whether S. aureus expresses a heme detoxification system in response to growth in heme, a proteomics-based technique known as two-dimensional difference gel electrophoresis (2D-DIGE) [24, 25] was utilized to search for proteins that change in abundance when S. aureus is exposed to heme. 2D-DIGE allows a quantitative comparison to be made of the abundance of individual proteins in a complex sample across multiple conditions. Following quantitative comparison of proteins between samples, individual proteins that change in abundance in a given condition can be identified by mass spectrometry.
Using 2D-DIGE, 21 proteins that display alterations in abundance when S. aureus is grown in high heme were identified [26]. Most of these proteins exhibit modest 2-to 5-fold changes in expression, however a single protein is increased in abundance an impressive 45-fold when S. aureus is grown in heme. This protein was identified by mass spectrometry as the ATPase component of an uncharacterized staphylococcal ABC transport system [23, 26]. Ubiquitous amongst bacteria and eukaryotic organisms, ABC transporters consist of an integral membrane permease and a peripherally associated ATPase that together couple the cleavage of ATP to the translocation of small molecules, ions, and proteins across the cell membrane [27–29]. In order to reflect the heme-responsive expression pattern of the ATPase component, this particular ABC transporter was named the heme-regulated ABC transporter (hrtAB; hrtA, ATPase; hrtB, permease) [23, 26]. HrtA and HrtB are predicted to be co-transcribed, an indication that they work together to form a functional transport system.
To test whether HrtAB plays a role in the previously described adaptive response of S. aureus to heme toxicity, mutants that lack the members of the hrtAB operon were generated [23]. These mutants were tested for the ability to adapt to growth in an otherwise toxic heme concentration by pre-exposure to heme and subculture into medium containing a high level of heme. These experiments revealed that mutants that lack either hrtA or hrtB fail to adapt to heme toxicity [23]. Furthermore, these mutants are highly sensitive to the bactericidal effect of heme on staphylococci. These findings indicate that HrtAB is critical for the ability of S. aureus to adapt to and proliferate in toxic concentrations of heme. Together with the heme-regulated expression of HrtAB, these data point to a model in which S. aureus senses heme and upregulates the expression of HrtAB, which results in relief from heme toxicity [23]. Given the predicted function of HrtAB as an ABC transporter, it was initially hypothesized that HrtAB exports excess heme that may accumulate in the staphylococcal cytoplasm due to the efficiency of heme uptake by the Isd and Hts systems. However, it is equally likely that HrtAB does not export heme itself from the cytoplasm, but exports byproducts of heme toxicity or heme metabolites that accumulate in bacterial cells upon heme acquisition, and thus alleviates heme toxicity through this mechanism. In support of this later contention, mass spectrometry-based tracking experiments using isotopically labeled heme have not revealed a role for HrtAB in staphylococcal heme efflux [unpubl. data].
During systemic infections, S. aureus is intimately associated with vertebrate blood, the most abundant source of heme in the host. Furthermore, the staphylococcal Isd and Hts systems have evolved to rapidly and efficiently acquire heme as a nutrient iron source. To cope with the toxicity inherent to the rapid intracellular amassing of heme, S. aureus relies on the heme-dependent upregulation of HrtAB to avoid heme toxicity [9, 10, 23, 30]. In keeping with this, it would appear likely that a mutant strain of S. aureus with impaired resistance to heme toxicity would have a diminished ability to cause disease as such a strain might succumb to heme toxicity during infection. However, S. aureus ΔhrtA displays a remarkable phenotype in a murine abscess model of systemic infection. Rather than being attenuated in its ability to infect mice and cause disease, S. aureus ΔhrtA exhibits increased virulence [23]. Mice infected with S. aureus ΔhrtA develop significant signs of disease early after infection and survive for a reduced length of time compared to mice infected with wild-type S. aureus. This increased virulence was further manifested upon autopsy, which revealed that infection with S. aureus ΔhrtA leads to considerable liver abscess formation and increased bacterial burden in this organ. Surprisingly, this hypervirulence appears to be liver-specific as examination of other organs in the same infection model does not reveal differences in virulence between S. aureus ΔhrtA and wild type.
A potential mechanistic explanation for the liver-specific hypervirulence of S. aureus ΔhrtA surfaced during comparison of the immune response to S. aureus wild type and ΔhrtA. Cell populations in the livers of mice infected with wild type and ΔhrtA were subjected to FACS-based analysis. These experiments revealed no appreciable strain-specific differences in the immune response to staphylococci colonizing the kidneys and spleens of infected mice. Furthermore, examination of infected livers did not reveal any differences in the levels of a number of cells of the innate immune system including natural killer cells, dendritic cells, and invariant natural killer T cells [23]. However, significant differences between livers from mice infected with wild type and ΔhrtA were observed in terms of the number of granulocytes and phagocytes present at the site of infection [23]. Specifically, livers of mice infected with ΔhrtA harbor half the number of CD11b+/CD11c− phagocytes and fewer CD11b+/Ly6G+ granulocytes than those from mice infected with wild type [23]. This is consistent with histological analysis of both sets of livers that revealed fewer polymorphonuclear cells present in tissue infected with ΔhrtA. These results suggest that S. aureus ΔhrtA either interferes with the recruitment of phagocytes and granulocytes to the site of infection or that ΔhrtA exhibits an increased ability to kill these cell types as they migrate toward invading staphylococci.
S. aureus is notorious for its ability to combat cells of the immune system through the secretion of effector molecules that interfere with leukocyte or lymphocyte function [31]. One possible explanation for the increased ability of S. aureus ΔhrtA to inhibit neutrophil recruitment to the site of infection is that when this heme-sensitive strain is under heme stress, it alters the quantity or the types of effector molecules that it secretes, thereby altering host cell function or survival. In order to test whether S. aureus ΔhrtA exhibits a heme-dependent alteration in the amount or types of secreted effector molecules, microarray and secreted protein profile experiments were performed on S. aureus ΔhrtA grown in the presence and absence of heme. These experiments revealed that upon exposure to heme, S. aureus ΔhrtA dramatically increases the expression and secretion of a number of immunomodulatory proteins that interfere with the recruitment of phagocytes to the site of infection [23, 30]. The secreted effector response of S. aureus ΔhrtA to heme stress seems to specifically inhibit the recruitment of host immune cells to the site of infection rather than increase the lysis of these cells, as the expression of a number of cytolytic exotoxins in this strain is repressed [30]. This secreted protein response correlates with the observation that fewer neutrophils and granulocytes are present in abscesses formed by S. aureus ΔhrtA, providing a possible explanation for the increased virulence of this strain [23]. These data are all consistent with a model in which S. aureus ΔhrtA encounters host heme during infection, which induces heme stress and causes this strain to secrete effector molecules that interfere with host phagocyte recruitment and function [23, 30]. This enables S. aureus ΔhrtA to form larger abscesses and reach higher levels that wild-type S. aureus in the liver.
It is unclear why the virulence of ΔhrtA is perturbed only in the livers of infected mice and not in other organs; however, it is possible that S. aureus encounters greater heme stress when infecting the murine liver than the kidney or spleen. In this regard, it can be envisioned that hemoglobin catalysis in the liver following normal erythrocyte turnover as a result of intravascular hemolysis increases the amount of heme available for interaction with invading staphylococci. It should be pointed out that the heme stress experienced by S. aureus within the host must be at a level significant enough to alter virulence gene expression but not so dramatic as to negatively impact growth of S. aureus ΔhrtA during infection. The fact that S. aureus ΔhrtA specifically increases the expression of immunomodulatory virulence factors in response to heme stress raises the tantalizing possibility that regulatory circuits exist within staphylococci that respond to stress and are capable of activating a secreted protein response that specifically targets immune cell recruitment [30].
The ABC transporter HrtAB has been demonstrated to be critical for the ability of S. aureus to proliferate in otherwise toxic heme levels [23, 26, 30]. Perturbation of this system also increases the virulence of this important human pathogen [23]. However, the original observation of a heme-dependent increase in HrtAB expression that led to the discovery of this system is a particularly important finding, suggesting the presence of a regulatory system that responds to heme and alters gene expression accordingly [26]. In order to identify potential candidates for an HrtAB-regulating system, the genomic context surrounding hrtAB in the S. aureus chromosome was investigated for genes encoding potential regulatory systems. This simple genomics-based approach was conceptually feasible because bacterial regulatory systems are often genomically co-localized with their cognate target genes [32].
Interrogation of the S. aureus chromosome for potential HrtAB-regulating systems revealed the presence of two open reading frames immediately adjacent to hrtAB that are predicted to be transcribed in the opposite direction from hrtAB [23]. Importantly, these genes are annotated as members of a bacterial two-component system (TCS). This TCS is predicted to be composed of a membrane-localized histidine kinase and a cytoplasmic response regulator with a DNA-binding domain. In general, membrane-localized histidine kinases recognize a particular environmental signal, which is thought to induce a conformational change in the protein, leading to autophosphorylation on a conserved histidine residue within the cytoplasmic domain of the histidine kinase [33]. Response regulators catalyze transphosphorylation from the histidine kinase they recognize to a response regulator aspartic acid residue, an event that increases the ability of these proteins to activate the transcription of target genes [34]. Interestingly, ABC transporter-TCS clusters have only been found in the Gram-positive Firmicutes and are generally thought to be involved in resistance to bacteriocins, which are antibacterial compounds made by one bacterial strain to inhibit the growth of other surrounding bacteria [32]. It is thought that bacteria that synthesize a particular bacteriocin encode a TCS that recognizes this compound, controlling a resistance response. However, another ABC transporter-TCS cluster in S. aureus (the GraRS/ApsRS TCS and the VraFG ABC transporter) has recently been found to be involved in the resistance of this organism to the toxicity of certain cell wall-active antibiotics as well as antimicrobial peptides [35–37]. These studies suggest that ABC transporter-TCS clusters may not be exclusively devoted to bacteriocin resistance, but may be involved in the resistance to toxic host compounds or antimicrobial agents.
The Heme Sensor System (HssRS)
Given the heme-regulated expression pattern of HrtAB and the fact that a TCS is found co-localized with hrtAB on the S. aureus chromosome, a model was envisioned in which this TCS responds to heme, thereby inducing the expression of HrtAB and initiating the S. aureus heme detoxification response. To test whether the hrtAB-proximal TCS is involved in the response of S. aureus to heme toxicity, mutant strains lacking either the histidine kinase or the response regulator gene were generated. As with S. aureus ΔhrtA, inactivation of either the response regulator or histidine kinase leads to an inability to adapt to heme toxicity or proliferate in high heme concentrations [23]. Furthermore, a strain of S. aureus lacking the hrtA-proximal response regulator displays an increased bacterial burden in the murine liver upon infection, a similar phenotype observed for S. aureus ΔhrtA [23]. This suggests that this TCS functions along with HrtAB to control the heme toxicity response of S. aureus during infection. This TCS was therefore designated the heme sensor system (hssRS; hssR, response regulator; hssS, histidine kinase) [23].
The fact that HssRS is required for the adaptation of S. aureus to heme toxicity as well as the predicted function of HssRS as a sensor system that regulates gene expression together imply that HssRS directly controls transcription of hrtAB. Three experiments were carried out to test this hypothesis. First, the level of HrtAB transcript was measured by RT-PCR in wild-type and S. aureus ΔhssR grown in the presence or absence of heme. In wild-type S. aureus, heme induces an increase in the level of HrtAB transcript, suggesting that the heme-regulated expression of HrtAB occurs at the level of gene expression [23]. This heme-dependent increase in the HrtAB transcript is not observed in a S. aureus strain lacking the hssR gene, implying that the HssRS TCS is critical for the ability of S. aureus to increase the expression of HrtAB in response to heme. Second, a plasmid-based reporter construct was generated to indirectly monitor HrtAB expression by fusing the hrtAB promoter to the coding sequence for the XylE reporter enzyme. Wild-type S. aureus harboring this hrtAB promoter-xylE fusion exhibits a significant heme-dependent increase in reporter activity, further indicating that the heme-dependent expression of HrtAB occurs through an increase in synthesis of the HrtAB transcript driven by the hrtAB promoter [23, 38]. Furthermore, no heme-dependent increase in reporter activity occurs in S. aureus ΔhssR or ΔhssS, establishing the importance of the HssRS TCS in the regulation of HrtAB expression in response to heme [23, 38]. Third, 2D-DIGE was utilized to determine the abundance of all of the S. aureus cytoplasmic proteins in wild type and ΔhssR grown in the presence and absence of heme [38]. This experiment revealed two important findings. First, HrtA is increased in abundance in response to heme in wild type but not in ΔhssR, further establishing the importance of HssRS in the regulation of HrtAB expression [38]. Second, HrtA is the only cytoplasmic protein that depends on hssR for a heme-dependent increase in abundance [38]. This suggests that the HssRS TCS may specifically regulate the expression of HrtAB rather than a set of genes [38].
These experiments demonstrate a dependence on HssRS for the response of S. aureus to heme toxicity through the regulation of HrtAB expression. However, they do not delineate the signaling events in S. aureus that connect HssRS to HrtAB expression. A series of experiments was therefore carried out to elucidate the mechanism by which HssRS regulates HrtAB expression in response to heme. Potential residues required for HssS-HssR signaling were identified by aligning the amino acid sequence of the predicted HssS cytoplasmic domain with the cytoplasmic domains from a number of known and characterized TCS histidine kinases. These experiments revealed a highly conserved histidine residue in HssS (His249) that aligns with the histidine residue known to be phosphorylated in response to signal recognition in other histidine kinases [38]. Similar alignments with HssR revealed a conserved aspartic acid residue (Asp52) that aligns with those residues known to undergo phosphorylation in other response regulators [38]. Based on the identification of these residues, it was postulated that upon signal recognition, HssS catalyzes autophosphorylation at His249 and that HssR subsequently catalyzes transphosphorylation to Asp52, activating the latter protein. To test this hypothetical signal transduction pathway, recombinant HssS and HssR as well as their predicted phosphotransfer site mutants were purified and tested for in vitro signaling. While HssS undergoes time-dependent autophosphorylation in these experiments, HssS mutated at its conserved histidine residue fails to undergo phosphorylation [38]. When HssR is added to phosphorylated HssS, HssR is able to catalyze transphosphorylation, indicating that these proteins are capable of phosphotransfer in vitro [38]. Furthermore, HssR mutated at its predicted phosphorylation site fails to catalyze transphosphorylation from HssS [38]. These experiments suggest that HssS undergoes autophosphorylation at His249 and that HssR catalyzes transphosphorylation to Asp52. However, these experiments were all carried out in vitro and therefore do not fully establish the importance of these signaling events in the functioning of HssRS in S. aureus.
To this end, the HssS and HssR mutants that fail to undergo signaling in vitro were tested for functionality in vivo. This was accomplished by creating plasmids containing a full-length copy of either wild-type HssS or HssS mutated at His249, which is required for in vitro autophosphorylation. Strains harboring these plasmids were tested for the ability to rescue the heme-sensitive phenotype of ΔhssS by growth curve analyses. Importantly, the plasmid bearing wild-type HssS rescues the growth of S. aureus lacking the hssS gene while the plasmid containing the mutant form of HssS that fails to undergo in vitro autophosphorylation is unable to rescue ΔhssS [38]. These differences occur in spite of significant expression of both proteins. Furthermore, a similar result is seen with HssR: while wild-type HssR rescues S. aureus lacking the hssR gene, HssR mutated at the aspartic acid residue required for in vitro phosphotransfer is unable to complement ΔhssR even though both proteins are expressed by S. aureus [38]. Cumulatively, these results suggest that HssS-HssR signaling is essential for the ability of S. aureus to respond to the toxicity of heme.
Given the observation that HssRS is required for an increase in transcription of hrtAB driven by the hrtAB promoter and the fact that HssR is modeled to contain a DNA-binding domain, it was predicted that HssR binds directly to the HrtAB promoter upon phosphorylation. To test this prediction, recombinant HssR was phosphorylated in vitro and added to magnetic beads coated with DNA corresponding to the hrtAB promoter. Proteins that associate with the hrtAB promoter are precipitated along with the magnetic beads used in this assay and can then be eluted from the beads and analyzed by immunoblot analysis. It was found that phosphorylated HssR is able to bind to such beads coated with hrtAB promoter DNA [38]. Importantly, phosphorylated HssR does not associate with beads coated with hrtAB intragenic DNA and non-phosphorylated HssR does not bind to the hrtAB promoter in vitro [38]. Furthermore, HssR mutated at its phosphorylation residue is not able to bind to the hrtAB promoter [38]. These experiments suggest that upon phosphorylation, HssR binds directly to the hrtAB promoter. To test the in vivo relevance of these observations, pull-down experiments using DNA-coated magnetic beads similar to those described above were performed using cytoplasmic extracts from S. aureus grown in the presence and absence of heme. These experiments revealed that HssR binds to the hrtAB promoter when S. aureus encounters heme, and that no binding of HssR to the hrtAB promoter occurs when S. aureus is grown in medium lacking heme [38]. Together, these results suggest that HssS senses heme and initiates a signaling pathway that leads to HssR phosphorylation and binding of phosphorylated HssR to the hrtAB promoter.
Response regulators such as HssR that contain DNA-binding domains usually exhibit specificity for certain sites within the promoters of the genes they regulate. Such sites often consist of short direct repeat sequences to which the effector domain of a response regulator is thought to associate through a head-to-tail dimerization mechanism [34]. Accordingly, the hrtAB promoter was interrogated for direct repeat sequences as a means of identifying potential HssR-binding sites. These analyses revealed a perfect direct repeat sequence upstream of the hrtAB coding sequence (GTTCATATT(N2)GTTCATATT) [38]. To test whether this direct repeat is involved in the response of the hrtAB promoter to heme-dependent activation by HssRS, the hrtAB promoter-xylE fusion reporter construct was subjected to mutational analyses. Truncation of the hrtAB promoter up to this direct repeat fails to alter activity of the hrtAB promoter, but truncation of half or all of this repeat completely eliminates hrtAB promoter heme responsiveness [38]. Furthermore, mutation of four residues within the hrtAB promoter that are conserved in the hrtAB promoters of other species of staphylococci also eliminates the response of this promoter to heme [38]. This suggests that the hrtAB promoter direct repeat is required for the ability of this promoter to respond to heme through HssRS. In light of the fact that HssR binds directly to the hrtAB promoter, in vitro promoter-binding experiments were carried out to test whether HssR binds to this direct repeat. To this end, HssR was eluted from the hrtAB promoter with increasing concentrations of double-stranded oligonucleotides corresponding to either the wild-type hrtAB promoter direct repeat or a pseudo-direct repeat mutated at the four residues required for the functioning of the hrtAB promoter in vivo. While the intact direct repeat elutes HssR, the mutated direct repeat fails to elute HssR from the hrtAB promoter [38]. These results suggest that upon phosphorylation, HssR binds to the direct repeat within the hrtAB promoter.
Together, the known features of HssS-HssR signaling and the mechanism by which these events impinge on the hrtAB promoter to regulate expression of HrtAB are shown in schematic form in figure 1. Nonetheless, a number of aspects of the HssRS and HrtAB systems remain to be elucidated. First, the transport substrate for HrtAB is unknown. Originally, it was proposed that HrtAB removes excess heme from the staphylococcal cytoplasm in order to alleviate heme toxicity [23, 30]. However, it is equally possible that a toxic molecule accumulates in S. aureus upon intracellular amassing of exogenously acquired heme. It can also be envisioned that breakdown of heme by S. aureus results in the buildup of toxic heme metabolites that must be exported from the staphylococcal cytoplasm. More studies focused on the function of HrtAB need to be carried in order to understand the mechanism of heme toxicity in S. aureus and the means by which HrtAB alleviates heme toxicity.
Fig. 1.
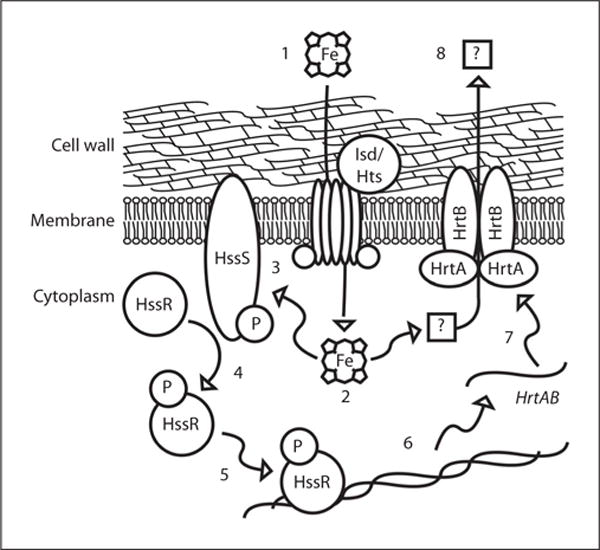
Signaling events that connect heme sensing by the HssRS TCS to upregulation of HrtAB and heme detoxification: (1) Heme is imported into the staphylococcal cytoplasm through the actions of the Isd and Hts systems. (2) Heme activates the histidine kinase HssS directly through a receptor-ligand interaction, or indirectly through the heme-mediated accumulation of toxic metabolites. (3) Activation of HssS results in autophosphorylation and (4) transphosphorylation of HssR. (5) Phosphorylated HssR binds to a direct repeat sequence within the hrtAB promoter. (6) This increases the transcription of the hrtAB genes by recruitment of RNA polymerase. (7) Newly synthesized HrtAB alleviates heme toxicity (8) through the export of heme or an unknown molecule that accumulates in the S. aureus cytoplasm upon heme exposure.
Another aspect of HssRS/HrtAB function that is not fully understood is the nature of the signal that is directly sensed by the histidine kinase HssS. Although HssS may sense heme by direct binding, HssS also could sense one of the toxic effects that heme has on the cell. Genomics-based approaches have not revealed a potential heme-binding domain in HssS, and the predicted sensing domain of this kinase has little sequence similarity to any protein with a known function [23]. Furthermore, no amino acids that typically engage in axial coordination of the iron atom of heme are conserved in the putative sensing domain of all of the potential HssS orthologues [unpubl. observations]. Identification of the ligand for HssS will lay the groundwork for a mechanistic understanding of how this histidine kinase is activated upon heme exposure. This would represent a significant advance for understanding HssS function and the mechanism employed by S. aureus to regulate the tight balance between heme acquisition and heme toxicity.
HssS represents one of the few histidine kinases that respond to a molecular marker of vertebrate tissue and one of two histidine kinases known to respond to heme. Importantly, other TCS that recognize heme are found in the Gram-positive pathogen Corynebacterium diphtheriae, a bacterium that is not closely related to S. aureus [39–42]. In C. diphtheriae, the TCS ChrAS and HrrAS are capable of responding to heme or hemoproteins [41]. However, in addition to the absence of any significant sequence identity between the staphylococcal and corynebacterial systems, two major aspects of ChrAS and HrrAS distinguish them from the S. aureus HssRS system. First, ChrAS and HrrAS both regulate expression of the C. diphtheriae heme oxygenase HmuO, while HssRS does not appear to regulate expression of the S. aureus heme oxygenases [38, 41, 42]. HmuO is capable of catabolizing heme and is therefore a critical factor required for heme metabolism in C. diphtheriae [43]. In S. aureus, IsdG and IsdI are iron-regulated heme oxygenases that also break down heme [10, 16, 44]. However, 2D-DIGE experiments have failed to reveal a role for HssRS in the regulation of IsdG/I expression, indicating that HssRS does not regulate these heme-degrading enzymes in S. aureus [38]. Second, despite the fact that HssRS from S. aureus and ChrAS/HrrAS from C. diphtheriae both respond to heme, the histidine kinase HssS and the kinases ChrS/HrrS differ dramatically in terms of the predicted structure of their signal-sensing domains. While HssS is predicted to have an N-terminal periplasmic sensing domain flanked by two transmembrane helices, ChrS/HrrS are predicted to have sensing domains composed of five transmembrane helices flanked by short loops [23, 42]. This difference in predicted structure suggests that HssS may sense heme through a mechanism that is different from that of ChrS/HrrS or that these TCS might sense different effects that heme has on the Gram-positive bacterial cell. Interestingly, C. diphtheriae strains lacking the chrAS genes exhibit elevated heme sensitivity that is not dependent on reduced HmuO expression [39]. This suggests the presence of a ChrAS-regulated heme-detoxification system in C. diphtheriae [39]. It is tempting to speculate that C. diphtheriae encodes a system functionally analogous to the S. aureus HrtAB system that protects this organism from heme toxicity.
The ability of bacterial pathogens to sense and respond to heme is not limited to Gram-positive organisms. The Gram-negative mammalian pathogen Bordetella pertussis also encodes a heme sensing system [45–51]. In B. pertussis, the heme receptor BhuR binds to heme, transducing signals into the periplasm that activate the cytoplasmic membrane protein RhuR [46, 48, 50]. RhuR alters the activity of the cytoplasmic protein RhuI, which regulates the expression of the B. pertussis heme uptake system BhuRSTUV [46, 48, 50]. Through this mechanism, B. pertussis is able to sense exogenous heme and increase the expression of systems required for heme uptake from the host. Thus the B. pertussis RhuIR/BhuR system differs from the HssRS system of S. aureus in that the former is devoted to the sensing of host heme for the purposes of heme acquisition, whereas the latter is required for sensing host heme to avoid heme toxicity [23, 38, 45–51]. Furthermore, HssRS is a TCS, while RhuI is an extracytoplasmic sigma factor and RhuR is a regulator of RhuI activity. Thus, the mechanism by which heme is sensed by these systems as well as the details of signaling are likely distinct. It appears that the pathogens S. aureus, C. diphtheriae and B. pertussis are all able to sense host heme and transduce signals that alter gene expression, but that these events occur through distinct mechanisms and for different reasons.
Evolutionary Implications of Heme Sensing in Gram-Positive Bacteria
A role for the TCS HssRS and the heme-regulated ABC transporter HrtAB has been established in the ability of S. aureus to avoid heme toxicity as well as in the pathogenesis of this organism [23, 38]. Interestingly, a number of distinct Gram-positive bacteria also encode potential orthologues of these systems (fig. 2) [23]. Most of these bacteria are either pathogens or saprotrophs (microorganisms that break down tissues of dead animals) and thus are likely to encounter host heme. For example, Bacillus anthracis, the cause of anthrax infection, is a bacterium that has a significant bloodstream component throughout the late stages of systemic infection [52]. B. anthracis also is capable of inducing hemolysis and has been shown to acquire iron from heme, potentially exposing this organism to heme toxicity during infection [53–56]. Accordingly, B. anthracis encodes potential HssRS/HrtAB orthologues (fig. 2) and has been shown to adapt to heme toxicity [23]. Although the putative B. anthracis Hss and Hrt systems have not been experimentally proven to be functional in this organism or orthologous to the corresponding systems in S. aureus, these initial observations suggest that B. anthracis must respond to heme toxicity at some point throughout its life cycle. Potential Hss and Hrt genes are also found in the pathogenic or saprotrophic Listeriae such as L. monocytogenes and L. innocua as well as other Bacilli including B. cereus and B. thuringiensis (fig. 2). Exiguobacterium sibiricum, an extremophile isolated from Siberian permafrost, also encodes potential Hss/Hrt systems (fig. 2). The details of the life cycle of this organism are currently unknown, but the fact that it encodes potential Hss/Hrt systems suggests that it may experience exogenous heme exposure in its natural habitat. Importantly, Hss and Hrt genes appear to be absent from the genomes of a number of non-pathogenic and non-saprotrophic Gram-positive bacteria including B. subtilis and B. licheniformis, a possible indication that Hss and Hrt are only required for the fitness of bacteria that encounter exogenous heme throughout their life cycles. The fact that Hss and Hrt genes are widespread throughout Gram-positive bacteria that associate with heme-containing tissues suggests that the ability of these organisms to overcome heme toxicity has been critical for their capacity to occupy different niches, whether pathogenic or saprotrophic. Furthermore, the fact that genes encoding Hss and Hrt are present in a number of human pathogens establishes the members of these systems as potential targets for therapeutic agents that could interfere with bacterial pathogenesis through the perturbation of heme metabolism. This idea is supported by the alteration in virulence observed upon inactivation of the staphylococcal Hrt systems.
Fig. 2.
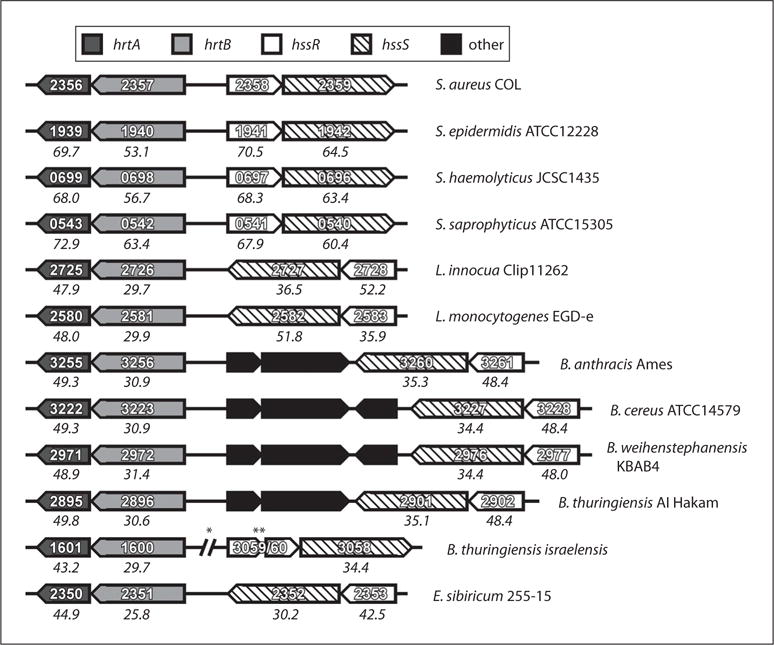
Conservation of HssRS and HrtAB across Gram-positive bacteria. Sequenced bacterial genomes were interrogated for potential orthologues of S. aureus hssRS and hrtAB genes by BLAST analyses. The indicated bacterial strains were found to encode potential Hss/Hrt systems. Boxes indicate open reading frames and point in the direction of transcription. Dark grey boxes: HrtA; light grey boxes: HrtB, empty boxes: HssR, boxes with diagonal lines: HssS; black boxes: non-Hss/Hrt open reading frames (other genes). Numbers within boxes indicate the annotated gene number. Numbers below boxes are percent similarities with respect to the corresponding S. aureus COL Hss/Hrt gene. Percent similarities were calculated at the amino acid level using a Lipman-Pearson Protein Alignment (gap penalty = 4, gap length penalty = 12). Single asterisk denotes large separation between genes; double asterisk denotes that this potential HssR orthologue is split into two open reading frames in the genome of this species.
Acknowledgments
We would like to thank members of the Skaar laboratory for reading of the manuscript. This work was supported by the Searle Scholars Program, United States Public Health Service Grant AI69233 from the National Institute of Allergy and Infections Diseases, and the Southeast Regional Centers of Excellence in Emerging Infections and Biodefense (SERCEB). E.P.S. holds an Investigator in Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. D.L.S. was supported by T32 HL069765 from the National Institute of Allergy and Infectious Diseases.
References
- 1.Fridkin SK, Hageman JC, Morrison M, Sanza LT, Como-Sabetti K, Jernigan JA, Harriman K, Harrison LH, Lynfield R, Farley MM. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352:1436–1444. doi: 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- 2.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 3.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 4.Creech CB, 2nd, Kernodle DS, Alsentzer A, Wilson C, Edwards KM. Increasing rates of nasal carriage of methicillin-resistant Staphylococcus aureus in healthy children. Pediatr Infect Dis J. 2005;24:617–621. doi: 10.1097/01.inf.0000168746.62226.a4. [DOI] [PubMed] [Google Scholar]
- 5.CDC. HIV/AIDS Surveillance Report. 2007:17. [Google Scholar]
- 6.Kuehnert MJ, Kruszon-Moran D, Hill HA, McQuillan G, McAllister SK, Fosheim G, McDougal LK, Chaitram J, Jensen B, Fridkin SK, Killgore G, Tenover FC. Prevalence of Staphylococcus aureus nasal colonization in the United States, 2001–2002. J Infect Dis. 2006;193:172–179. doi: 10.1086/499632. [DOI] [PubMed] [Google Scholar]
- 7.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 8.Creech CB, 2nd, Talbot TR, Schaffner W. Community-associated methicillin-resistant Staphylococcus aureus: the way to the wound is through the nose. J Infect Dis. 2006;193:169–171. doi: 10.1086/500432. [DOI] [PubMed] [Google Scholar]
- 9.Skaar EP, Schneewind O. Iron-regulated surface determinants (Isd) of Staphylococcus aureus: stealing iron from heme. Microbes Infect. 2004;6:390–397. doi: 10.1016/j.micinf.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Reniere ML, Torres VJ, Skaar EP. Intracellular metalloporphyrin metabolism in Staphylococcus aureus. Biometals. 2007;20:333–345. doi: 10.1007/s10534-006-9032-0. [DOI] [PubMed] [Google Scholar]
- 11.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 12.Bullen JJ, Griffiths E. Iron and Infection: Molecular, Physiological and Clinical Aspects. New York: Wiley; 1999. [Google Scholar]
- 13.Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 14.Crichton R. Inorganic Biochemistry of Iron Metabolism: From Molecular Mechanisms to Clinical Consequences. Chichester: Wiley; 2001. [Google Scholar]
- 15.Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O. Passage of heme-iron across the envelope of Staphylococcus aureus. Science. 2003;299:906–909. doi: 10.1126/science.1081147. [DOI] [PubMed] [Google Scholar]
- 16.Skaar EP, Gaspar AH, Schneewind O. IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J Biol Chem. 2004;279:436–443. doi: 10.1074/jbc.M307952200. [DOI] [PubMed] [Google Scholar]
- 17.Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP. Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J Bacteriol. 2006;188:8421–8429. doi: 10.1128/JB.01335-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O. Iron-source preference of Staphylococcus aureus infections. Science. 2004;305:1626–1628. doi: 10.1126/science.1099930. [DOI] [PubMed] [Google Scholar]
- 19.Graca-Souza AV, Maya-Monteiro C, Paiva-Silva GO, Braz GR, Paes MC, Sorgine MH, Oliveira MF, Oliveira PL. Adaptations against heme toxicity in blood-feeding arthropods. Insect Biochem Mol Biol. 2006;36:322–335. doi: 10.1016/j.ibmb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Tolosano E, Fagoonee S, Hirsch E, Berger FG, Baumann H, Silengo L, Altruda F. Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood. 2002;100:4201–4208. doi: 10.1182/blood-2002-04-1270. [DOI] [PubMed] [Google Scholar]
- 21.Stojiljkovic I, Evavold BD, Kumar V. Antimicrobial properties of porphyrins. Expert Opin Investig Drugs. 2001;10:309–320. doi: 10.1517/13543784.10.2.309. [DOI] [PubMed] [Google Scholar]
- 22.Francis SE, Sullivan DJ, Jr, Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 23.Torres VJ, Stauff DL, Pishchany G, Bezbradica JS, Gordy LE, Iturregui J, Anderson KL, Dunman P, Joyce S, Skaar EP. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe. 2007;1:109–119. doi: 10.1016/j.chom.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman DB. Quantitative proteomics for two-dimensional gels using difference gel electrophoresis. In: Matthiesen R, editor. Mass Spectrometry Data Analysis in Proteomics. Totowa/NJ: Humana Press; 2006. [Google Scholar]
- 25.Lilley KS, Friedman DB. All about DIGE: quantification technology for differential-display 2D gel proteomics. Expert Rev Proteomics. 2004;1:401–409. doi: 10.1586/14789450.1.4.401. [DOI] [PubMed] [Google Scholar]
- 26.Friedman DB, Stauff DL, Pishchany G, Whitwell CW, Torres VJ, Skaar EP. Staphylococcus aureus redirects central metabolism to increase iron availability. PLoS Pathog. 2006;2 doi: 10.1371/journal.ppat.0020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 28.Pohl A, Devaux PF, Herrmann A. Function of prokaryotic and eukaryotic ABC proteins in lipid transport. Biochim Biophys Acta. 2005;1733:29–52. doi: 10.1016/j.bbalip.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Young J, Holland IB. ABC transporters: bacterial exporters-revisited five years on. Biochim Biophys Acta. 1999;1461:177–200. doi: 10.1016/s0005-2736(99)00158-3. [DOI] [PubMed] [Google Scholar]
- 30.Stauff DL, Bagaley D, Torres VJ, Joyce R, Anderson KL, Kuechenmeister L, Dunman PM, Skaar EP. Staphylococcus aureus HrtA is an ATPase required for protection against heme toxicity and prevention of a transcriptional heme stress response. J Bacteriol. 2008;190:3588–3596. doi: 10.1128/JB.01921-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. 2005;3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 32.Joseph P, Fichant G, Quentin Y, Denizot F. Regulatory relationship of two-component and ABC transport systems and clustering of their genes in the Bacillus/Clostridium group, suggest a functional link between them. J Mol Microbiol Biotechnol. 2002;4:503–513. [PubMed] [Google Scholar]
- 33.Mascher T, Helmann JD, Unden G. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol Mol Biol Rev. 2006;70:910–938. doi: 10.1128/MMBR.00020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao R, Mack TR, Stock AM. Bacterial response regulators: versatile regulatory strategies from common domains. Trends Biochem Sci. 2007;32:225–234. doi: 10.1016/j.tibs.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M. Gram-positive three-component antimicrobial peptide-sensing system. Proc Natl Acad Sci USA. 2007;104:9469–9474. doi: 10.1073/pnas.0702159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol Microbiol. 2007;66:1136–1147. doi: 10.1111/j.1365-2958.2007.05986.x. [DOI] [PubMed] [Google Scholar]
- 37.Meehl M, Herbert S, Gotz F, Cheung A. Interaction of the GraRS two-component system with the VraFG ABC transporter to support vancomycin-intermediate resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51:2679–2689. doi: 10.1128/AAC.00209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stauff DL, Torres VJ, Skaar EP. Signaling and DNA-binding activities of the Staphylococcus aureus HssR-HssS two-component system required for heme sensing. J Biol Chem. 2007;282:26111–26121. doi: 10.1074/jbc.M703797200. [DOI] [PubMed] [Google Scholar]
- 39.Bibb LA, King ND, Kunkle CA, Schmitt MP. Analysis of a heme-dependent signal transduction system in Corynebacterium diphtheriae: deletion of the chrAS genes results in heme sensitivity and diminished heme-dependent activation of the hmuO promoter. Infect Immun. 2005;73:7406–7412. doi: 10.1128/IAI.73.11.7406-7412.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitt MP. Transcription of the Corynebacterium diphtheriae hmuO gene is regulated by iron and heme. Infect Immun. 1997;65:4634–4641. doi: 10.1128/iai.65.11.4634-4641.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitt MP. Identification of a two-component signal transduction system from Corynebacterium diphtheriae that activates gene expression in response to the presence of heme and hemoglobin. J Bacteriol. 1999;181:5330–5340. doi: 10.1128/jb.181.17.5330-5340.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bibb LA, Kunkle CA, Schmitt MP. The ChrA-ChrS and HrrA-HrrS signal transduction systems are required for activation of the hmuO promoter and repression of the hemA promoter in Corynebacterium diphtheriae. Infect Immun. 2007;75:2421–2431. doi: 10.1128/IAI.01821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitt MP. Utilization of host iron sources by Corynebacterium diphtheriae: identification of a gene whose product is homologous to eukaryotic heme oxygenases and is required for acquisition of iron from heme and hemoglobin. J Bacteriol. 1997;179:838–845. doi: 10.1128/jb.179.3.838-845.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu R, Skaar EP, Zhang R, Joachimiak G, Gornicki P, Schneewind O, Joachimiak A. Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J Biol Chem. 2005;280:2840–2846. doi: 10.1074/jbc.M409526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirby AE, Metzger DJ, Murphy ER, Connell TD. Heme utilization in Bordetella avium is regulated by RhuI, a heme-responsive extracytoplasmic function sigma factor. Infect Immun. 2001;69:6951–6961. doi: 10.1128/IAI.69.11.6951-6961.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.King ND, Kirby AE, Connell TD. Transcriptional control of the rhuIR-bhuRSTUV heme acquisition locus in Bordetella avium. Infect Immun. 2005;73:1613–1624. doi: 10.1128/IAI.73.3.1613-1624.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy ER, Sacco RE, Dickenson A, Metzger DJ, Hu Y, Orndorff PE, Connell TD. BhuR, a virulence-associated outer membrane protein of Bordetella avium, is required for the acquisition of iron from heme and hemoproteins. Infect Immun. 2002;70:5390–5403. doi: 10.1128/IAI.70.10.5390-5403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirby AE, King ND, Connell TD. RhuR, an extracytoplasmic function sigma factor activator, is essential for heme-dependent expression of the outer membrane heme and hemoprotein receptor of Bordetella avium. Infect Immun. 2004;72:896–907. doi: 10.1128/IAI.72.2.896-907.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanderpool CK, Armstrong SK. The Bordetella bhu locus is required for heme iron utilization. J Bacteriol. 2001;183:4278–4287. doi: 10.1128/JB.183.14.4278-4287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanderpool CK, Armstrong SK. Heme-responsive transcriptional activation of Bordetella bhu genes. J Bacteriol. 2003;185:909–917. doi: 10.1128/JB.185.3.909-917.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brickman TJ, Vanderpool CK, Armstrong SK. Heme transport contributes to in vivo fitness of Bordetella pertussis during primary infection in mice. Infect Immun. 2006;74:1741–1744. doi: 10.1128/IAI.74.3.1741-1744.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mock M, Fouet A. Anthrax. Annu Rev Microbiol. 2001;55:647–671. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 53.Shannon JG, Ross CL, Koehler TM, Rest RF. Characterization of anthrolysin O, the Bacillus anthracis cholesterol-dependent cytolysin. Infect Immun. 2003;71:3183–3189. doi: 10.1128/IAI.71.6.3183-3189.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skaar EP, Gaspar AH, Schneewind O. Bacillus anthracis IsdG, a heme-degrading monooxygenase. J Bacteriol. 2006;188:1071–1080. doi: 10.1128/JB.188.3.1071-1080.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaspar AH, Marraffini LA, Glass EM, Debord KL, Ton-That H, Schneewind O. Bacillus anthracis sortase A (SrtA) anchors LPXTG motif-containing surface proteins to the cell wall envelope. J Bacteriol. 2005;187:4646–4655. doi: 10.1128/JB.187.13.4646-4655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maresso AW, Chapa TJ, Schneewind O. Surface protein IsdC and Sortase B are required for heme-iron scavenging of Bacillus anthracis. J Bacteriol. 2006;188:8145–8152. doi: 10.1128/JB.01011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]