

Abstract
Age, apolipoprotein E ɛ4 (APOE) and chromosomal sex are well-established risk factors for late-onset Alzheimer’s disease (LOAD; AD). Over 60% of persons with AD harbor at least one APOE-ɛ4 allele. The sex-based prevalence of AD is well documented with over 60% of persons with AD being female. Evidence indicates that the APOE-ɛ4 risk for AD is greater in women than men, which is particularly evident in heterozygous women carrying one APOE-ɛ4 allele. Paradoxically, men homozygous for APOE-ɛ4 are reported to be at greater risk for mild cognitive impairment and AD. Herein, we discuss the complex interplay between the three greatest risk factors for Alzheimer’s disease, age, APOE-ɛ4 genotype and female sex. We propose that the convergence of these three risk factors, and specifically the bioenergetic aging perimenopause to menopause transition unique to the female, creates a risk profile for AD unique to the female. Further, we discuss the unique risk of the APOE4 positive male which appears to emerge early in the aging process. Evidence for impact of the triad of AD risk factors is most evident in the temporal trajectory of AD progression and burden of pathology in relation to APOE genotype, age and sex. Collectively, the data indicate complex interactions between age, APOE genotype and gender that belies a one size fits all approach and argues for a precision medicine approach that integrates across the three main risk factors for Alzheimer’s disease.
Keywords: Age, ApoE, sex difference, Alzheimer’s disease, mitochondria, bioenergetics, brain
1. Introduction
The greatest risk factors for Alzheimer’s disease are age(1–3), the ApoE4 allele(4–6) and female sex (1, 7–10). Prevalence of AD is greater in women (1) whereas the incidence for AD is reported to be comparable in women and men (11) until later age when the incidence is greater in women (12) (Figure 1). Typically, the greater risk of AD in females is attributed to their greater longevity of, on average, 4.5 years.
Figure 1.

Sex-specific incidence estimates of Alzheimer’s per 1,000 person years. Obtained with data from the Cache County Study [12]. Data reported in Ruitenberg et al., [12] indicate that men are at greater risk than women for developing earlier onset AD. However, this sex difference is reversed by age 75, with women at a 2 fold greater risk for AD, thereafter.
Herein, we review the three greatest risk factors for AD and propose that it is the interaction between these risk factors that either increase or decrease risk of Alzheimer’s. The complexity of the interaction between this triad of risk factors belies simple on factor causality but instead more closely approaches the complexity of factors leading to late onset Alzheimer’s disease (LOAD). While this approach takes into consideration three factors that have broad systems biology impact, it still does not fully capture the complexity of the processes leading to risk of LOAD. However considering the interaction between the three greatest risk factors for AD is a step in the right direction.
1.1. Aging
Age remains the greatest risk factor for Alzheimer’s and is thus a fundamental driver for development of the disease(13). Aging at the global population level is unprecedented and without parallel in the history of humanity(14). During the 20th century the proportion of older persons continued to rise and this trend has continued into the twenty-first century(14). By 2050, the number of older persons in the world will exceed the number of young for the first time in history(14). Between 2015 and 2030, the number of people in the world aged 60 years or over is projected to grow by 56% and by 2050 the global aged population of is projected to more than double(15). Globally, the number of people aged 80 years or over, the “oldest-old” persons, is growing the fastest. People 80 or older currently constitute more than 3% of the population of Northern America and nearly 3% of the population of Europe. Population ageing is a pervasive global phenomenon that will endure for the foreseeable future. By 2050, one in every five people will be aged 60 years or over(15).
The female prevalence of AD is well documented and is generally attributed to the greater life span of women relative to men(1). Globally, women outlive men by an average of 4.5 years(15). However, survival of males is projected to be comparable to females with near equality in longevity between females and males(15). Thus, the greatest risk factor for AD will be equally distributed between the sexes in the near future. Data from the multiple studies show that the incidence of Alzheimer’s disease rises exponentially after the sixth decade of life (16).
Aging is a complex progressive process involving every organ and cell system in the body and is the result of coordinated systems biology events that can span decades(17–27). Further, aging is typified by a coordinated series of sequential steps involving specific pathways at each phase in the aging process (17, 28). Systems either driving brain aging are contributors to risk of AD and include glucose hypometabolism and mitochondria dysfunction, innate immune and inflammatory reactions, beta amyloid processing, dysregulation of cholesterol homeostasis, white matter degeneration and decline in regenerative capacity (16, 17, 19, 21, 22, 24–26, 29, 30)
While aging is typically described and depicted as a linear process, in reality the program of senescence is nonlinear and better represented as step functions between transition states. Aging is typified by transition states and because aging is occurring throughout multiple organ systems the process of aging is highly complex(17, 19–21, 24). Each phase of the aging process can be considered a transition state that can be modified to either accelerate or delay progression to the next phase of aging (17, 22, 25).
It is becoming increasingly clear that complex systems have critical thresholds, often referred to as tipping points, when a system shifts from one state to the other(17). Analyses of transition state dynamics predict three health states, healthy, pre-disease and disease(17). The pre-disease transition state is typically defined as the limit of the normal state that precedes the tipping point into disease. Importantly, the pre-disease state is unstable and is thus potentially reversible. However, the duration of reversibility is limited. The bifurcation point between pre-disease and disease is characterized by a critical slowing of the system, in which it becomes increasingly slow to recover from small perturbations to equilibrium(17).
Transition states are inherently unstable and in the case of neurological transition states, indicators of dysfunction at the limits of normal can be signals of instability and tipping points. The presence, variability, intensity and duration of neurological symptoms provide potential advance warning signs of impending risk of later health risks, particularly neurodegenerative diseases. Multiple conditions that emerge during aging, such as metabolic dysregulation, cholesterol dyshomeostasis, insomnia, depression, subjective memory complaints and cognitive decline are associated with increased risk of neurodegenerative diseases later in life such as Alzheimer’s(17, 31). Alzheimer’s, like multiple neurodegenerative diseases, is characterized by a long prodromal period, 20 years, during which the disease progresses to clinically diagnosed dysfunction. Identifying persons that are at risk for AD while still in a modifiable transition states is critical for reversing or delaying development of Alzheimer’s (31).
Individual genetics form the foundation for and can modify the complex multidimensional trajectory of aging (32). Considered below are the two major genetic risk factors of AD, APOE genotype and chromosomal sex.
2. Overview of APOE
2.1. Biological Role and Evolution of APOE
Apolipoprotein E (ApoE) is a 34-kDa lipid binding protein that functions in the transport of triglycerides and cholesterol in multiple tissues, including brain, by interacting with lipoprotein receptors on target cells (33, 34) (1–4). ApoE is a cholesterol transporter and functions as a key regulator to coordinate the mobilization of cholesterol between cells and to redistribute cholesterol within cells. These functions are particularly critical for the nervous system where ApoE transport of cholesterol is critical for maintenance of myelin and neuronal membranes both in the central and peripheral nervous systems (35). In the CNS, ApoE functions in conjunction with APOJ and APOC1, which together deliver cholesterol necessary for membrane remodeling, required for synaptic turnover and dendritic reorganization (35). ApoE is particularly critical to the brain as other cholesterol transporters abundant in the plasma, such as ApoA1 and ApoB, are virtually absent in brain thus making the brain particularly reliant upon ApoE for cholesterol transport (35).
APOE, a3.6 kb long gene, is located on chromosome 19 and(36) encodes for apolipoprotein E (apoE), a 299 amino acid long lipoprotein. Three ApoE isoforms exist in humans: ApoE2, ApoE3, and ApoE4, which differ from one another by single amino acid substitutions at positions 112 and 158, ApoE2 (Cys-112, Cys-158), ApoE3 (Cys-112, Arg-158), and ApoE4 (Arg-112, Arg-158) (5). Substitution of cysteine at position 158 in ApoE2 results in hypocholesterolemia caused by low levels of low-density lipoprotein (LDL), cholesterol(5). In contrast, substitution of cysteine with arginine at position 112 in ApoE4 results in elevation of plasma cholesterol and LDL levels and predisposes the carrier to cardiovascular disease and neurodegenerative disorders, including Alzheimer’s disease (AD) (5).
APOE evolved from the common hominid ancestor of humans and the great apes [9]. While there are three main isoforms of APOE in humans (i.e. ɛ2, ɛ3, and ɛ4), all great apes carry the APOE-ɛ4 allele(37). The ɛ3 allele is the most common in humans, especially in regions with a long-established agricultural economy. However, the ancestral ɛ4 allele remains high in regions where an economy of foraging still exists or where food-supply is often scarce(38). Although generally more common among African populations, the risk in cognitive decline conferred by carrying the ɛ4 allele is greater among individuals of European descent, particularly amongst northern European regions (39).
2.2. APOE Protein Structure: Lipoprotein and Beta Amyloid Binding Motifs
As depicted in Figure 2, the ApoE protein has an N-terminal receptor-binding region and C-terminal hydrophobic lipid-binding region located at amino acids 244–272. It is the N-terminal region, which contains the two polymorphic positions (i.e. positions 112 and 158) that distinguish the differing isoforms in many ways, such as differing antioxidant profiles (where ɛ2 ≥ ɛ3> ɛ4) (40, 41). In APOE-ɛ4, Arginine-61 interactions with Arginine-112 lead to a conformational change causing Arginine-61 to interact with Glutamine-255 in the aqueous environment, resulting in N- and C-terminal domain interactions that do not exist in the ɛ2 or ɛ3 isoforms, due to the less hydrophobic Cysteine-112 (42). These interactions are particularly evident when apoE is delipidated, preventing lipoprotein receptor docking and internalization of unlipidated apoE. APOE-ɛ4 also has an arginine-112 to glutamic acid-109 salt bridge, which causes the arginine-61 side chain to extend away from the 4-helix bundle in its structure. In turn, the arginine-61 domain interacts with glutamic acid-255 causing the structure of ɛ4 to become more compact compared to ɛ3. Structural differences lead to regions in ɛ4 that are less protected and less stable than the same regions in ɛ3 (43). This difference also leads to a binding profile that favors triglyceride-rich very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL) over small phospholipid-rich high-density lipoproteins (HDL) particles. Compared to the structure of ɛ3, ɛ2 also causes changes in lipoprotein preference, due to impaired lipolytic processing of VLDL, creating increases in VLDL and decreases in LDL cholesterol (44). These differences in the ɛ2 and ɛ4 structures lead to increases in pro-atherogenic lipoproteins, an accelerated trajectory towards atherogenesis compared to ɛ3, and therefore an increased representation in patients with hyperlipidemia and cardiovascular disease (45). However, despite this high prevalence and the strength of its association with both cardiovascular and cognitive risk, APOE testing is still used most exclusively for research purposes only.
Figure 2.
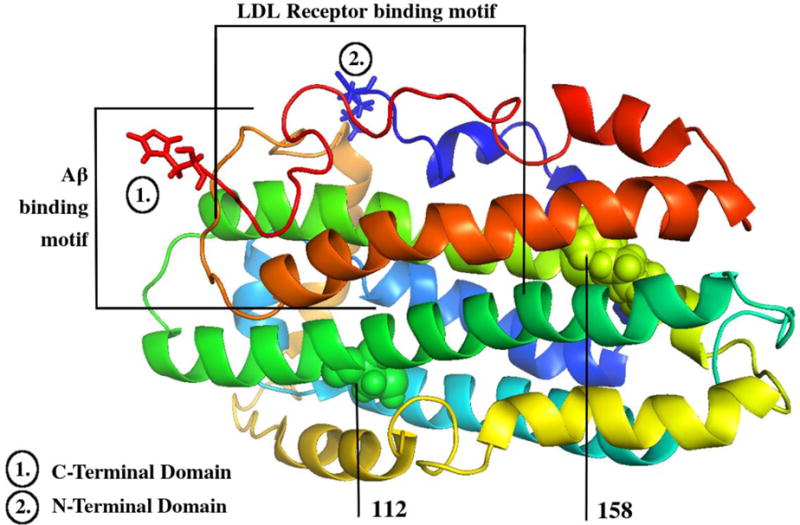
ApoE NMR protein structure. Created from the RCSB Protein Databank and the PyMol graphics viewer (PDB ID: PO2649) [186]. This figure shows the nuclear magnetic resonance protein structure of apoE ɛ3. Lipoprotein binding (1) and Aβ-binding (2) motif regions are delineated [34, 78]. Residues 112 and 158, which are altered between (and therefore determine) the differing isoforms, are also labeled [42–43].
2.3 ApoE and Cholesterol Transport
The APOE gene is expressed most highly in the liver and brain (34, 35, 46). Over 75% of plasma apoE protein is derived from hepatic synthesis (46). Second to the liver, is brain synthesis of apoE which predominantly occurs in astrocytes, followed by microglia and neurons (47). Given this high production, apoE is the most prevalent brain lipoprotein. The level of APOE expression varies by genotype, with ɛ2 typically having the greatest and ɛ4 having the lowest expression (48, 49). APOE isoform effects on plasma apoE concentration accounts for 20% of inter-individual variability of apoE concentration across all ages, with other factors including BMI, waist-to-hip ratio, oral contraceptives, and sex, also playing prominent role (50).
APOE isoforms differ in their rates of catabolism such that ɛ2 is catabolized more slowly than ɛ3 or ɛ4 (51). The increased rate of catabolism of apoE 4 leads to reduced availability of apoE to serve as a clearance protein, required for the clearance of cholesterol and toxic amyloid-beta (Aβ) oligomers (52).
Excess plasma apoE stimulates hepatic VLDL production and impairs lipolysis whereas a low level of apoE impairs plasma clearance of triglyceride-rich lipoproteins. Accordingly, there is about a 3-fold increase in AD-risk associated with the lowest tertile of apoE plasma concentrations, independent of APOE genotype (53). Notably, apoE plasma levels are lowest in MCI patients that progress to AD. Given that ɛ2 carriers generally show the highest protein concentration, there is some level of reduction in AD-risk in these individuals, while ɛ4 carriers have an increased risk. Studies of cultured astrocytes show that apoE-ɛ3 containing astrocytes are able to release more cholesterol to supply neurons than apoE-ɛ4 containing astrocytes (54).
ApoE-ɛ4 protein is also less efficient at delivering essential fatty acids such as docosahexaenoic acid (DHA) to neurons, thereby altering the function of glucose transporters and possibly playing a role in the development of insulin resistance, a state commonly attributed to an increased risk of AD (55). Given this alteration, it has been suggested that eiscosapentaenoic acid (EPA)-rich oils may be more suitable in ɛ4 individuals, especially for use as a hypotriglyceridaemic agent, showing a greater hypercholesterolaemic response to DHA than EPA (56). Indeed, there is a significant interaction between APOE genotype and plasma lipid response to both diet and pharmacological therapies (57). Since a ketogenic diet has shown to increase HDL and decrease LDL cholesterol in both obese and normal weight individuals, it has been proposed that ɛ4 carriers would benefit more from a ketogenic diet than non-carriers, particularly in regards to protection from AD (58–60). Furthermore, diets higher in saturated fats or diets rich in olive oil and high in monounsaturated fats both show more favorable outcomes in LDL-particle count in ɛ4 individuals than ɛ3 individuals (61–64); LDL-particle count is seen as a better measure of atherogenicity than LDL cholesterol itself (65, 66).
Due to the altered protein structure, APOE-ɛ4 is poorly lipidated by ABCA1 and ABCG1, causing it to be more rapidly catabolized than the other isoforms. Furthermore, APOE polymorphisms show a differential impact on competition between fasting triglyceride-rich lipoproteins and LDL’s for the LDL receptor (LDL-R) (67), ultimately leading to downregulation of LDL-R in states of high VLDL production, as seen in carriers of the ɛ2 and ɛ4 allele. These LDL-R interactions are necessary for remnant clearance of the partially metabolized atherogenic chylomicrons and VLDL particles. Interestingly, APOE-ɛ4 in non-human primates does not contain the domain interactions that gives human ɛ4 its characteristic salt-bridge, thus it behaves more like ɛ3 in humans in that it does not show preferential binding to triglyceride-rich lipoproteins like the human variant (68). In line with this, it has been proposed that gene targeting to replace arginine-61 with threonine-61 would likely correct the lipid binding preference that leads to the toxic effects of ɛ4 (69).
The cholesterol efflux regulatory protein (CERP), also known as ABCA1 (ATP-binding cassette transporter), is a major regulator of cholesterol and phospholipid transport and homeostasis. ABCA1 is promotes lipidation of apoE, which allows it to efficiently bind Aβ and facilitate Aβ uptake through LRP1. Studies of APP mice with an ABCA1 deficiency have increased amyloid deposition in the brain, paralleled by decreased levels of apoE. Microdialysis assays indicate that ABCA1 deficiency significantly decreases the clearance of Aβ in apoE-ɛ4-expressing mice, while there are no significant effects of ABCA1 on Aβ clearance in apoE-ɛ3-expressing mice (6).
Liver X receptor (LXR) is a nuclear receptor transcription factor that plays an essential role in regulation of cholesterol, triglycerides, fatty acids, and glucose homeostasis. Bexarotene, which activates LXR/retinoid X receptor (RXR) heterodimers, increases the expression of apoE, reduces amyloid plaques, and improves memory performance in an AD mouse model harboring the ɛ4 allele (70). Likewise, treatment with natural LXR agonists, such as the algae derived Fucosterol has shown to induce transcriptional activation of apoE, and the cholesterol efflux regulator and HDL mediator, ABCA1, without increasing cellular accumulation of triglycerides, a side-effect of other LXR agonists (59).
LXR agonists have also been shown to increase the activities of mitochondrial mediated antioxidant enzymes, such as SOD and glutathione (71).
Polymorphisms in the Translocase of the Outer Mitochondrial Membrane (TOMM40) gene, which is in linkage disequilibrium with APOE, are associated with late-onset AD risk and age of disease onset (72). The expression of both apolipoprotein E and TOMM40 are significantly increased with AD. APOE expression and TOMM40 expression mRNA levels in the temporal and occipital cortexes are reportedly higher in carriers of the very long TOMM40 variant (523 poly-T), compared to those with the short variant, both in normal individuals and those with AD. This increased TOMM40 expression leads to an increased risk of AD in ɛ3/ɛ4 carriers and not ɛ3/ɛ3 carriers (72). However, it is unclear what this means in terms of TOMM40 protein levels. For instance, although both show alteration associated with AD pathology, mRNA of APOE expression does not always correlate with apoE protein levels (73). An additional variant at the TOMM40/APOE gene cluster (rs2075650) has been significantly associated with LDL and C-reactive protein (CRP) levels, with a trend toward association with HDL and triglyceride levels, proteins that have all been associated with modulation of risk for heart disease and AD (73). Beyond TOMM40, most genes involved in the APOE gene cluster (i.e. APOC1, APOC2, and APOC4) have been associated with risk for AD. For instance, apoC2, which serves as a measure of triglyceride rich lipoproteins, has been found to co-localize with Aβ (74, 75). Similar findings of increased triglycerides with increased apoC1 have been found, along with strong associations with risk of dementia and insulin resistance (76, 77)
Taken together these findings indicate that there are structural and widespread metabolic differences between the APOE isoforms. These widespread differences play a prominent role in conferring risk for Alzheimer’s.
2.4. ApoE, Beta Amyloid and Tau
APOE is the best-characterized amyloid-β (Aβ) chaperone, with apoE containing an Aβ-binding motif around region 230–243 (see Figure 1) (34, 78). Functional APOE can induce intracellular Aβ degradation through trafficking of amyloid to lysosomes. However, the three isoforms differ in their binding affinities. Specifically, the direct interaction between Aβ and apoE facilitates Aβ oligomerization in rank order ɛ4> ɛ3> ɛ2(79, 80).
As lipidated forms of APOE-ɛ4 bind poorly to soluble Aβ, it is suggested that lipoproteins may contribute in other ways to the dynamics of Aβ clearance along the cerebral vasculature(81). Indeed, APOE enables cerebrovascular integrity through the cyclophilin A-NFκB-MMP pathway. Astrocytes in ɛ3 mice suppress the cyclophilin A-NFκB-MMP pathway, while in ɛ4 mice they do not, indicating the ɛ3 isoform may have greater vascular protective capabilities (82). Further, APOE knockout mice experience progressive blood-brain-barrier (BBB) breakdown (83).
APOE-ɛ4 has been shown to increase Aβ deposition and Aβ oligomer formation. Additionally, studies of PiB binding, a technique used to image amyloid, have shown that ɛ4 increases brain amyloid burden in a dose-dependent manner in cognitively normal individuals and individuals in the prodromal stages of AD (84, 85). Furthermore, ɛ4 is associated with greater membrane disruption and lysosomal leakage in the presence of Aβ compared to ɛ3. Interestingly, reductions in glucose metabolism have been shown to correlate better with APOE genotype than amyloid load, showing that genotype contributes to reduced glucose metabolism in aging independently of Aβ. Indeed, APOE-ɛ4 individuals show less variation in CSF Aβ levels from a cognitively normal status to mild cognitive impairment (MCI), believed to be a precursor to AD, than do non-carriers (86).
Mice with an ɛ4 allele insertion show increased tau accumulation, the main component of neurofibrillary tangles seen within somatodendritic and intra-axonal compartments in those diagnosed with AD (87). Moreover, apoE-ɛ3 is able to bind tau to prevent tau phosphorylation and assembly into paired-helical filaments, while ɛ4 is not (88). Additionally, ɛ4 carriers consistently show higher tau, p-tau, and tau/Aβ-42 ratios (89). Although these associations are present in both sexes, females show a greater association with CSF tau, pointing to possible sex differences along the disease trajectory (10). Future studies involving the use of the lipid-probe theta-toxin, which binds cholesterol, in 3xTgAD and App-NL-F mice, may help further elucidate the sex interactions between apoE and AD pathogenesis (90).
Under conditions of oxidative and excitotoxic stress or neuronal damage, apoE is produced by neurons (47). Due to the domain interaction of ɛ4, when apoE is synthesized in this state it undergoes proteolytic cleavage to a much greater extent than ɛ3 (91), with resulting truncated C-terminal fragments entering the cytosol where they become neurotoxic (92) as well as toxic to mitochondria (93). Importantly, during oxidative phosphorylation, mitochondria are one of the primary sources of oxidative stress [92–93]. In line with this, post-mortem microarray analysis of brains of middle-aged cognitively normal APOE-ɛ4 and APOE-ɛ3 carriers without pathological evidence of AD have revealed differences in gene transcripts that are associated with both mitochondrial function and AD, including complex-I gene NADH dehydrogenase, as well as genes associated with insulin signaling, such as CTNNB1 (94). Interestingly, no differences in transcripts directly involved in amyloid processing were found between the groups. Support for the early role of mitochondrial bioenergetics in the progression of AD is strengthened by evidence that young ɛ4 carriers show reductions in cytochrome-c oxidase (COX) activity, a measure of oxidative metabolism, in brain tissue from the posterior cingulate, even in the absence of fibrillar amyloid (95). Likewise, AD patients with the ɛ4 allele show higher levels of hydroxyl radicals than AD patients without an E4 allele and reduced levels of glutathione peroxidase, an enzyme with antioxidant capacity that is produced in mitochondria (71, 96). These differences may partially be explained in that mass spectrometry analysis have revealed the methionine-108 region of apoE is considerably more reactive toward free radical labeling in ɛ4 (97). APOE-ɛ4 carriers also have higher levels of isoprostanes, a marker of oxidative stress, than non-ɛ4 carriers when matched for cholesterol levels (98). Finally, decreases in mitochondrial respiratory complexes are seen in neurons of ɛ4 mice and not ɛ3. In fact, treatment of these cells with GIND25, a small molecule that disrupts the detrimental ɛ4 structural domain interaction has shown to restore mitochondrial respiratory complex IV levels to a level similar to ɛ3 neurons (94). A similar compound, IAH, is currently being studied to elucidate its therapeutic potential in AD patients (99). These findings support the early role of energy metabolism in the progression of AD, and suggest that compromises in mitochondrial dysfunction may precede plaque formation in ɛ4 carriers. It is likely that amyloid fibrils later amplify this mitochondrial dysfunction.
Taken together, these findings indicate that the structural differences in apoE between the isoforms result in a cascade of physiological effects. These differences result in both Aβ-dependent and Aβ-independent changes that serve to effect overall risk for Alzheimer’s. In the ɛ4 isoform, these changes include an increased avidity to bind pro-atherogenic lipoproteins, alterations in the bioenergetic profile, reduced ability for apoE to clear Aβ, and increased accumulation of tau. Furthermore, the apoE ɛ4 isoform leads to an increased production of reactive oxygen species (ROS) and therefore increased neurotoxic and toxic mitochondrial environments under conditions of oxidative stress.
2.5. APOE-ɛ4 Specific Risks of Alzheimer’s Disease
Among persons diagnosed with AD, up to 60% carry at least one ɛ4 allele (8). APOE ɛ4 exerts its maximal effect on AD-risk by the early 70’s (100), with a reduction in risk after age 85 in both sexes (8). Similarly, the accelerating effect of ɛ4 on rates of decline diminishes with advancing disease stages, such that APOE genotype has no significant effect on overall disease duration. The APOE-ɛ4 allele accounts for as much as 50% of the genetic attributable AD risk (8, 101). In persons diagnosed with AD, APOE-ɛ4 homozygotes carriers are diagnosed at a mean age 68 years whereas in ɛ4 heterozygotes mean age is 76 years, and 84 years in ɛ4 noncarriers (6). The APOE ɛ4 gene dose effect on risk and age of AD onset indicates that APOE ɛ4 dramatically increases risk of AD and an earlier age of onset.
In women, carrying one APOE ɛ4 allele shifts the AD risk curve five years earlier, while two alleles shifts the curve to 10 years earlier in both women and men (102) (Table 1). In addition, the risk odds ratio for AD in women with one allele is 3.5–4, while it is 12–15 in APOE ɛ4 homozygote women and men (103, 104).
Table 1.
Summary of the results outlined in this review. Both protective and risk factors associated with APOE genotype by sex are outlined.
| Summary of Sex Differences by APOE Genotype | ||
|---|---|---|
| Genotype | Sex | Findings |
| ɛ2/ɛ2 | Males | More severe lipidemia and atherosclerosis due to high plasma apoE [64] |
| Females | Reduced AD risk in women <85, increased after age 85 [166] | |
| General | Increased prevalence of hyperlipidemia and cardiovascular disease [45] ApoE concentrations are generally the highest in this group, protecting from AD [48, 53] |
|
| ɛ2/ɛ3 | Males | Increased AD risk due to higher incidence of insulin resistance [64] |
| Females | Estrogen and ERT improve insulin sensitivity, compensating for the increased risk in males [64] Reduced cardiovascular risk and AD risk in women <85, increased after age 85 [64, 166] |
|
| General | Associated with increased longevity and protection from AD [161–165] | |
| ɛ3/ɛ3 | Males | Potentially protective against AD, although at higher risk than genotype-matched females [121] |
| Females | MCI patients show lowest levels of p-tau, associated with delayed disease progression [121] More favorable CSF amyloid profiles than ɛ3/ɛ3 men [121] Lowest AD risk compared to males and all other APOE genotypes in females [121] Increases in apoE concentration with age provide protection from AD risk [121, 48] Associated with increased longevity and protection from AD [161–165] |
|
| General | Protective against AD [for example: 121, 127, 39, 110, 69, 162, 166] | |
| ɛ2/ɛ4 | Males | Increased risk of cardiovascular disease compared to ɛ3/ɛ3 individuals [64] |
| Females | Protection from cardiovascular risk and AD [121, 64, 166] | |
| General | Greater AD risk in individuals of European descent [39] Evidence of brain amyloid accumulation occurs 10–15 years later than in ɛ4/ɛ4 carriers [127] |
|
| ɛ3/ɛ4 | Males | Increased risk for MCI and AD, although slower decline [121] |
| Females | Significant effects on risk for decline in episodic memory [180] Higher MCI and AD risk than non-ɛ4 carriers and male ɛ3/ɛ4, comparable to ɛ4/ɛ4 men [7, 121] Increased likelihood of pathological levels of CSF tau and tau/Aβ ratio [121] Compared to ɛ3/ɛ4 men, faster age-related cognitive decline and longer survival rates [8, 109, 124] |
|
| General | In 70+ normal ɛ4 carriers have greater amyloid PET than other genotypes [127] Greater AD risk in individuals of European descent [39] Evidence of brain amyloid accumulation occurs 10–15 years later than for ɛ4/ɛ4 carriers [127] |
|
| ɛ4/ɛ4 | Males | Higher risk of MCI and AD than ɛ3/ɛ4 or ɛ2/ɛ4 males or ɛ3/ɛ4 and ɛ4/ɛ4 females [8, 121, 166] |
| Females | Comparable AD risk to ɛ3/ɛ4 females [6]; longer survival rates following AD diagnosis [7,8] | |
| General | Over 30% develop AD by age 75, with greater risk in individuals of European descent [39] ApoE concentrations are generally the lowest in this group, a predictor of AD risk [48, 53] Highest risk for brain Aβ accumulation [84–85] |
|
| General ɛ4 findings | Males | Years of life lost in men ɛ4 carriers are greater than in genotype-matched women [167] |
| Females | Greater prevalence of AD in female ɛ4 carriers than males [7, 8] Greater alterations in precuneus and anterior cingulate cortex connectivity [10, 125] Associations between APOE and cognitive decline evident later in females [9, 127] More prominent phenotypic features in female MCI ɛ4 carriers – reduced hippocampal volume and worse cognitive scores [126] CSF tau, p-tau, and tau/Aβ-42 levels highest in MCI ɛ4 carriers compared to non-carriers with MCI, particularly for females [7, 10, 12, 89] Women ɛ4 with mild-AD are more likely to have both neurofibrillary tangles and amyloid than ɛ4 men, indicating greater pathology [128] Postmenopausal ɛ4 women on ERT exhibit signs of neuroprotection and preservation of telomere length compared to ɛ4 women not receiving treatment [130, 144, 150] |
|
| General |
APOE-ɛ4 show greater neural efficiency on episodic memory tasks [105] and better performance in speed of processing, attention, and verbal fluency until mid 50’s when declines are evident [64, 106–107] Reduced myelin integrity in AD-relevant regions evident in infancy [152] Increased dementia risk (AD, PDD, VaD, and DLB) and cardiovascular risk [12, 64, 121] Carrying one ɛ4 allele shifts the AD risk curve 5 years earlier (OR=3.5–4) [102–104] Two alleles shifts the AD risk curve by 10 years (OR=12–15) [102–104] APOE-ɛ4 exerts its maximal effects on AD risk by the early 70’s [100] Up to 65% of individuals with AD harbor at least one ɛ4 allele [8] CSF Aβ less predictive of conversion from normal to MCI than for other genotypes [86] By 40, 15% cognitively normal APOE-ɛ4 carriers are amyloid positive [127] |
|
Aβ: Amyloid-beta 1–42; AD: Alzheimer’s disease; DLB: Dementia with Lewy Bodies; ERT: Estrogen replacement therapy; MCI: Mild cognitive impairment; OR: Odds ratio; PDD: Parkinson’s dementia; P-tau: Phosphorylated tau; VaD: Vascular dementia.
APOE may serve as an interesting example of antagonistic pleiotropy – a gene that confers advantage in one period of life but later presents as disadvantage. Specifically, young individuals with the ɛ4 allele show greater neural efficiency on tasks of episodic memory as measured through a more rapid decline in blood-oxygen-level dependent (BOLD) response over learning trials and therefore more efficient use of memory resources (105). They also show better performance in speed of processing, attention and verbal fluency (106). This cognitive benefit extends into middle-age (i.e. 45–55) on measures of attention and prospective memory (107). However, this benefit does not extend to verbal memory at this age. In fact, a study comparing individuals 45–57, and 58–68, and age-matched controls found that the 45–57 apoE-ɛ4 group performed better on verbal memory tests than the control group, while the 58–68 group showed impairments. At this later age, ɛ4 older adults also show increased fMRI activation in the association cortex during memory challenge and during tasks involving verbal fluency tests compared to non-ɛ4 age-matched controls (108, 109), as well as altered connectivity in the default mode network. Further, stressful life events have greater impact on cognition of ɛ4 individuals of Caucasian-American descent compared to African Americans (110).
Despite the potential early benefits, older ɛ4 individuals show evidence of early reduction in entorhinal cortex volume (111), greater rate of hippocampal volume loss (112), and early rise in rate of myelin breakdown(113) (Table 1). These individuals also show characteristic patterns of parietal, cingulate, and temporal hypometabolism, consistent with an AD-like pattern in brain glucose utilization (114–116). When stratifying by APOE genotype, positron emission tomography (PET) imaging is predictive of AD conversion with 100% sensitivity and 90% specificity (117). Conversely, APOE ɛ2 carriers show evidence of protective mechanisms in the brain, particularly in regards to white matter compared to ɛ3 carriers (Table 1). Specifically, they exhibit higher fractional anisotropy, a measure of fiber integrity, and lower radial diffusivity, a measure of myelin integrity, in the posterior cingulate and anterior corpus callosum, regions relevant to the progression of AD (118, 119).
3. Sex Differences in APOE Genotype and Risk of Alzheimer’s
3.1. Evidence for Worse Pathology and Accelerated Degeneration Rates in Females relative to Males: Impact of APOE-ɛ4
Postmenopausal women constitute >60% of the affected Alzheimer population and are those who will bear the greatest burden of the disease (1, 2, 120). Twenty years, 2 decades, ago Farrer and colleagues reported a sex difference in the lifetime risk of AD in women (8). In his seminal report, women with a single copy of the ApoE4 allele was sufficient to increase disease risk associated with two copies of the ApoE4 gene in men (8). This finding was confirmed a year later in a subsequent report by Payami, Schellenberg and colleagues (7) who found that ApoE4 heterozygote men had lower risk than ApoE4 homozygotes; there was not a significant difference between epsilon4 heterozygote males and those without epsilon4 (Table 1). In contrast, epsilon 4 heterozygote women had the same significant twofold increased risk as homozygote men (7). Multiple studies indicate that the APOE-ɛ4 risk for AD is greater in women, especially in heterozygous individuals carrying one APOE-ɛ4 allele (7, 8, 10, 121). Women with one copy of the APOE-ɛ4 allele have a 4-fold increase in the risk of AD, (7, 8, 121) whereas women and men with two copies of the ApoE4 allele exhibit a 15-fold increase in risk (7, 8, 121) and a significantly lower age of onset compared with AD patients carrying ApoE2 or 3 alleles. Consistent with greater AD risk, Barnes, Bennet and colleagues found a even greater sex difference in the impact of pathology and risk of AD 6. Each additional unit of AD pathology was associated with a nearly 3-fold increase in the odds of clinical AD in men compared with a more than 22-fold increase in the odds of clinical AD in women (122).
Women diagnosed with mild cognitive impairment progressed at faster rates of cognitive decline than men (123). Further, women had an acceleration rate of cognitive decline relative to men. The gender effect was greatest in ApoE4 female carriers. Overall, women ɛ3/ɛ4 carriers often show faster age-related decline and greater deterioration of cognition than elderly ɛ3/ɛ4 men (8). Indeed, as measured by rates of atrophy and clinical presentation, MCI women decline more rapidly (109) while at the same time experiencing a longer survival rate following diagnosis in ɛ3/ɛ4 and ɛ4/ɛ4 women (124).
In a study of 131 participants, decreased connectivity in the default mode network in healthy older APOE-ɛ4 carriers was identified, with a significant sex interaction in the precuneus, a major hub for the default mode. Specifically, female ɛ4 carriers show significantly reduced connectivity compared to female ɛ3 carriers and male ɛ4 carriers in the anterior cingulate cortex. Additional studies have revealed that cognitively normal female ɛ4 carriers show greater reduction of functional connectivity in the precuneus compared to ɛ4 males. This region is structurally connected to the medial temporal lobe, the initial site of tau pathology in AD. This region also shows reduced glucose metabolism in early AD and asymptomatic ɛ4 carriers (125). Furthermore, MCI female ɛ4 carriers show more prominent phenotypic features than their male counterparts, such as reduced hippocampal volumes and worse cognitive scores (126). Similar to the differences in cognitively normal ɛ4 carriers, female ɛ4 MCI patients show decreased precuneus activity compared to non-ɛ4 women and ɛ4 men. In MCI, this decreased connectivity extends to the posterior cingulate (125).
By the age of 40, 15% of ɛ4 homozygous cognitively normal individuals are amyloid positive, a frequency which occurs 10–15 years earlier than for ɛ2/ɛ4 or ɛ3/ɛ4 individuals. By 90, more than 80% of ɛ4 are amyloid positive, although no sex differences are present (127). Given the lack of sex differences in amyloid by ɛ4, this supports the role of tau in mediating sex differences. In line with this, female ɛ4 carriers with MCI show greater CSF tau and tau/Aβ ratios compared to ɛ4 males with MCI (7). Similarly, ɛ4 women with mild AD are at a higher risk of having both neurofibrillary tangles and Aβ plaques than ɛ4 men with mild AD (128). These findings support the idea that women ɛ4 carriers have a greater extent of AD pathology as measured by CSF and pathology analysis at autopsy. While other studies have failed to find a sex difference in AD risk by APOE genotype, this can be explained in that there appears to be a differential effect on risk by carrying one ɛ4 allele compared to individuals who are homozygous for ɛ4, such that women with one allele are at a greater risk compared to ɛ3/ɛ3 women, while men have no increased risk with just one ɛ4 compared to ɛ3/ɛ3 men.
3.2. APOE Genotype and Response to Estrogen
On average, ApoE ɛ4 negative women have better cognitive performance than ApoE ɛ4 positive women(129). Further, ApoE ɛ4 positive women were at greater risk for cognitive impairment (129). The impact of estrogen therapy in postmenopausal women is complex. ApoE ɛ4 negative women receiving estrogen or hormone therapy had the highest level of cognitive performance, whereas women positive for ApoE ɛ4 receiving estrogen or hormone therapy performed worse than ApoE ɛ4 carriers not receiving therapy (129). Notably, treatment with tamoxifen, a selective estrogen receptor modulator and estrogen receptor antagonist, ameliorated cognitive deficits observed during the menopausal transition, especially in ApoE ɛ4 women (130).
The contradictory effects of estrogenic agonists and an estrogen receptor antagonist in ApoE ɛ4 carriers suggest that the female ApoE ɛ4 brain is different that the ApoE 3 carriers. We propose that one fundamental difference between the ApoE ɛ4 brain and Apoe 2 and 3 brains is in reliance of the ApoE ɛ4 brain on ketone bodies as a bioenergetic fuel. Reliance of the ApoE ɛ4 brain on ketone bodies as a bioenergetic fuel puts that brain at risk for utilization of its own white matter as fuel (29). In the female brain, estrogen activates the system biology of glucose metabolism while simultaneously suppressing the ketogenic system in brain thereby promoting brain reliance on glucose as its primary fuel to generate ATP (17, 28, 131–138). If the ApoE ɛ4 brain is a dual fuel dependent brain, being dependent upon glucose and ketone bodies, then suppression of the ketogenic system in the ApoE ɛ4 brain would result in reduction in a critical fuel to generate ATP. In the case ApoE 2 and 3 carriers, estrogenic activation of the glucose metabolism system and suppression of the ketogenic system is beneficial as estrogen is promoting and sustaining brain utilization of the primary fuel system glucose. In contrast, in a dual fuel dependent brain, one dependent upon both glucose and ketone bodies, suppression of the ketogenic pathway would put that brain at metabolic risk. Reliance of the ApoE ɛ4 brain on ketone bodies as a bioenergetic fuel is consistent wtih evidence from multiple laboratories demonstrating that ApoE ɛ4 carriers are glucose hypometabolic prior to decline in cognitive function (117, 139, 140).
Estrogen receptors play a key and contrasting role in regulation of ApoE gene and protein expression (33) and risk of AD(141). ERα up-regulated ApoE mRNA and protein expression whereas in contrast, ERβ down-regulated ApoE mRNA and protein expression(33). These data suggest that use of ER-selective ligands could provide therapeutic benefit to reduce the risk of AD by increasing ApoE expression in ApoE 2 or 3 allele carriers and decreasing ApoE expression in ApoE4 allele carriers. Polymorphisms in both estrogen receptor alpha (rs4986938) and estrogen receptor β (rs2234693) have been associated with increased risk of dementia and AD (141).
Estrogen therapy may prove beneficial in APOE-ɛ4 women who show a favorable response by increasing ABCA1 production. In fact, estrogen has been shown to increase ABCA1 mRNA expression in mice as well as postmenopausal women receiving hormone therapy (142, 143). Competitive interactions between estrogen receptor β and LXRβ for transcriptional coactivator RAP250 likely modulate estrogens effects on ABCA1 levels (143).
Postmenopausal APOE-ɛ4 positive women age 49–69 who discontinued their hormone therapy regimen exhibited telomere shortening, an index of biological aging, to a greater extent than women who do not carry an ɛ4 allele or ɛ4 women not receiving hormone therapy (144).
Estrogen exposure of mitochondria is implicated in regulation of mitochondrial functioning and activates manganese superoxide dismutase (MnSOD) antioxidant activity (145). Conversely, APP and Aβ disrupt mitochondrial function and this is partially mediated by Tom40 interactions (146). The Tom40 protein is an active channel for protein sorting, and is crucial to healthy mitochondria (146). As previously mentioned, the TOMM40 gene is in linkage disequilibrium with APOE (72). Notably, estrogen treatment has been shown to increase both apoE and Tom40 levels through activation of estrogen receptors (147). Estrogen treatment also modulates the apoE receptor LDL related receptor protein (LRP1) (148). Estrogen treatment in ovariectomized mice also causes increases in LRP, an apoE binding protein, in the hippocampus and neocortex (149). Along these lines, apoE synthesis is required for estrogen-induced neuroprotection and neurite outgrowth (148, 150, 151) and is lost in the presence of ɛ4 (151).
Collectively, these data indicate a complex interaction between the estrogenic and ApoE systems that deserve greater investigation at the systems biology level of analysis and particularly at the interaction between these two systems during nature aging.
3.6. Lack of Sex Differences in Young May Support a Role of Hormone Loss During Menopause
Infants harboring the APOE-ɛ4 allele have lower gray matter volume and lower white matter myelin water fractions, indicating reduced myelin integrity in AD-relevant regions compared to non-carriers. These include the precuneus, posterior cingulate, lateral temporal, and occipitotemporal regions (152). These infants also present with greater water fraction and gray matter volume in frontal regions, with no observable sex differences. Interestingly, they have greater myelin water fraction in regions that myelinate later and reduced water fraction in regions that myelinate early, potentially indicating a condensed white matter development trajectory. As AD is first associated with myelin loss in regions that myelinate late in development, a reduction in white matter development would put APOE-ɛ4 carriers at greater risk. In line with this, differences in APOE genotypes are associated with a steeper rate of myelin breakdown in late-myelinating frontal regions, and APOE has shown to mediate myelin maintenance and repair mechanisms (153). While there are observable differences in brains of APOE-ɛ4 carriers quite early in development, the impact of sex on APOE related changes in white matter or gray matter are not always found in studies looking at children or adolescents (111, 152). This evidence suggests that sex differences conferring greater risk of AD in ɛ4 women could be synergistic with events that occur during mid-life or later.
Interestingly, apoE concentration shows sex-specific alterations consistent with the time-scale of puberty, as well as menopause in women (see Figure 3). Specifically, females show higher apoE concentrations than males until around age 17, the age of puberty completion in women (154, 155). These differences are then reversed around the average age of menopause (48, 156). Importantly, this perimenopausal transition is marked by a bioenergetic shift in the brain to utilizing ketone bodies as fuel (17, 60, 157) with estrogen loss post-menopause resulting in decreased metabolic function in the brain (158–160).
Figure 3.
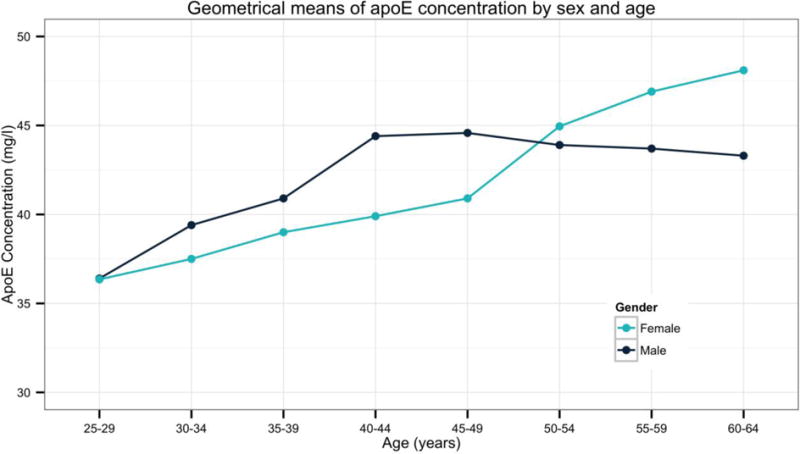
ApoE serum concentrations by sex and age (N=6,934). From data collected in the ApoEurope project, obtained with permissions from [48]. Data indicate that men have greater apoE concentrations than women until age 50–54. While men experience progressive declines in apoE concentrations following this age, apoE concentrations rise in women. As low serum levels of apoE are associated with an increased risk for AD, this tipping point might explain the discordant findings in risk by sex, such that men are at risk due to a reduced production of apoE, while ɛ4 women produce an overabundance of as isoform with impaired function.
3.7. Evidence for Protection of ɛ2/ɛ2 and ɛ3/ɛ3 Genotypes in Women
Over 87% of centenarians are ɛ2/ɛ3 or ɛ3/ɛ3 among the majority of populations studied, including France, Japan, Spain, Italy, and Finland (161–164). While the ɛ3 allele is the most common, the prevalence among this age group is greater than the general population. Notably, interactions with ACE polymorphisms, shown to impact cardiovascular risk, likely impact the role of APOE in promoting longevity (165). Although ɛ2 is generally believed to be protective against AD, evidence exists to suggest this is true in ɛ2/ɛ3 individuals of both sexes, but only in ɛ2 homozygous females [110; 134]. In line with the role of ɛ2 in promoting longevity, one study of Swedish individuals found that ɛ2 was associated with a reduced risk of AD only in females until age 85, when the allele stops being protective (166). Additionally, there is a decreased mortality rate in ɛ2 women, while this is not always observed in men (167). However, it is important to note that carriage of the ɛ2 allele is associated with a greater risk for hyperinsulinemia and diabetes in both sexes, both of which increase the risk for Alzheimer’s (168, 169).
The APOC1 gene, which is contained in the APOE gene cluster, has also been associated with longevity. Specifically, one SNP in this gene (rs4420638) has been identified as protecting against AD and promoting resilience. Interestingly, this SNP is in linkage disequilibrium with APOE and shows sex specific differences (170). In fact, APOC1 allele and genotype frequencies have been identified as significantly different in elderly women age 84 and older compared to younger women (171).
CSF analysis of MCI patients revealed higher levels of total tau in female ɛ4 carriers, and lowest levels of phosphorylated tau in ɛ3 females over ɛ3 males, potentially pointing to a protective factor in ɛ3 females over males (121). Additionally, cognitively normal ɛ3 homozygote females had higher CSF amyloid than ɛ3 homozygous males (121). Further, apoE serum concentrations continue to rise with age in ɛ3 women to a greater extent than ɛ4 women or ɛ3 men (48) (Figure 3). Given the role of APOE in Aβ sequestration and clearance, this increase likely provides for protection in ɛ3 women (121). Unfortunately, most studies looking at APOE genotype effects on the brain and CSF levels group all non-ɛ4 carriers together, making it difficult to more adequately assess whether or not the ɛ3 allele is protective in homozygous women.
In the latest study of over 8,000 individuals by Altman and colleagues comparing the risk of ɛ4 status by sex, an increased relative risk for female ɛ4 carriers (both heterozygous and homozygous) was identified over female ɛ3 homozygotes in progressing from normal cognition to MCI, or from MCI to AD. The absolute risk values were higher for male ɛ4 carriers with 22.4% of ɛ4 heterozygous male carriers progressing from normal cognition to MCI, whereas 19.8% of female ɛ4 heterozygous carriers did (121). Rather than indicating female ɛ4 carriers are at a greater risk, what this might indicate is that the impact of having an ɛ4 is greater in females, likely because there is some protective mechanism for the ɛ3 females. Indeed, the ɛ3 females showed the lowest risk of all groups. Specifically, only 13.9% of ɛ3 homozygous females progress from normal cognition to MCI, while 20.7% of males in this same category progress; additionally, 24.2% of females progress from MCI to AD, while 28% of males progress, indicating protection in the female ɛ3 carriers (121). Likewise, there is a higher proportion of male APOE ɛ3 homozygotes than females that have progressed to AD in the NACC Uniform Data Set, a database which involves data collected from 34 AD centers across the US (N=11,654) (172, 173).
3.8. Sex Differences in Cardiovascular Risk by APOE Genotype and Effects on Vascular Dementia
In a Framingham Heart Study of 3413 participants, age-adjusted period prevalence of cardiovascular disease (CVD) was related to APOE genotype with a higher rate for men than women (18.6% in the ɛ4 group for men and 9.9% for ɛ4 women) (64). Notably, while the ɛ2 allele was protective for women (4.9%), this protection was not seen in males with the ɛ2 allele (18.2%), indicating that the presence of either the ɛ2 or ɛ4 alleles in men is associated with greater CVD risk. Furthermore, the overall odds of CVD for men within the ɛ2 group was 1.94-fold greater than that for men within the ɛ3 group, while ɛ2 women were at a .91 reduced risk compared to the ɛ3 women (64). In a separate Framingham Heart Study of 7901 participants, mortality due to cardiovascular disease that was 6 times higher in men than women at ages 45–54 (174). The increased period prevalence in risk for heart disease for men was still evident when comparing the lowest risk group of males (i.e. ɛ3/ɛ3) to the highest risk group of women (i.e. ɛ4/ɛ4). However, after the age of 65, the sex difference in risk between males and females was less than 2-fold.
The incidence for vascular dementia has been shown to be higher in men than women (12). In fact, results from a recent study looking at sex differences by APOE genotype, cannot rule out that more vascular dementia pathologies could have been present in the male participants. While female ɛ4 carriers showed greater tau levels than ɛ4 males with MCI, the differences are in line with a potential vascular dementia in the males, as vascular dementia presents with similar CSF Aβ but lower total tau and phosphorylated-tau levels than AD (12). Importantly, participants presenting with potential vascular disease are often not included in studies of AD. While this is beneficial in that it limits potential confounds, there is also a growing understanding in the connection between AD and cerebrovascular health. Given that men are at a greater risk for heart disease and other cardiovascular events, especially towards the young-old age spectrum (e.g. 65–74), understanding this group would shed light on the sex differences in ɛ4 carriers.
Although cardiovascular deaths in late mid-life cannot explain all differences in dementia rate by sex, a substantial part of the difference in dementia risk between females and males could derive from incidence of cardiovascular disease before the age of 65. At mid-life lifetime risk of AD, is not different for men and women whereas after midlife for women and men and subsequent to menopause in women, from 65 years of age onward, women have a 2 fold greater lifetime risk of AD then men. While one could speculate that lower risk of AD in males over the age of 65 is due to survivor cohort. This hypothesis will be testable at the population level as males gain longevity comparable to females in the near future(15).
Men with ɛ2/ɛ3 alleles exhibit higher insulin levels compared with controls, indicative of a state of insulin resistance that has been associated with impaired clearance of atherogenic triglycerides. In women, estrogen therapy has been shown to improve insulin sensitivity, potentially providing protection prior to menopause (64). Men also show greater levels of SREBP2 expression, a protein that interacts with apoE to further regulate lipid homeostasis (175). Moreover, estrogen can upregulate apoE gene expression by increasing apoE mRNA (147). In fact, female rats also show higher expression levels than males (176). Given the impact of estrogen on apoE levels, this higher expression would allow for greater protection in terms of both cognition and heart health in females until menopause, when estrogen levels decline dramatically. This increase likely proves protective in ɛ2 women against CVD, while detrimental in ɛ4 women towards the risk of AD due to the pathological structure and function of ɛ4.
3.9. Evidence for Worse Effects in Men or No Sex Difference
Most intervention studies that include a separate analysis of possible therapeutic benefit in ɛ4 individuals, fail to consider that carrying an ɛ4 allele is associated with an earlier age of diagnosis and accelerated pathology. Therefore, age-matched studies often yield negative results in ɛ4 individuals. Similarly, AD shows an altered trajectory in time towards diagnosis between the sexes (see Figure 2), (177) likely blurring the sex interactions in ɛ4 carriers. For instance, the association between APOE genotype and cognitive decline is significant only in women over the age of 70 (9). Additionally, ɛ2 participants are often excluded from analysis, though studies that have included them indicate that having the ɛ2/ɛ4 allele is associated with an increased risk for AD compared to ɛ3/ɛ3 [19; 59; 134]. Given the differential impact this genotype has on women and men for cardiovascular health, it is likely that surviving men would exhibit an increased risk for AD with this genotype compared to females (178). However, there is likely to be a selection pressure on men ɛ4 carriers, such that those that overcome the prominent impact of this allele on cardiovascular risk likely possess other factors that compensate for the presence of an otherwise ‘frail’ genotype.
Low plasma apoE is associated with a decrease in hippocampal volume, and shows less of a sex difference in concentration levels in ɛ4 individuals than between the other genotypes (179). However, normal ɛ4 men show greater AD-pathology and worse memory performance than normal ɛ4 women, pointing to an earlier incidence of vulnerability (127). Notably, years of life lost in individuals with at least one ɛ4 allele are 2.5-fold greater for men than for ɛ4 women (167). Furthermore, there is some evidence of a sex by APOE gene dose interaction, such that the significant effects of ɛ4 on risk for decline in episodic memory are generally less dependent of zygosity in females than males (180). Likewise, ɛ4/ɛ4 men are at higher risk of AD than ɛ4/ɛ4 women (8). For example, in a recent study, a greater proportion of ɛ4/ɛ4 men progressed from normal cognition to a state of MCI than did ɛ4/ɛ4 women. Similar trends were seen for individuals starting as MCI at baseline and progressing to AD (121). Additionally, episodic memory as measured through the Kendrick Object Learning Test has been reportedly worse in normal ɛ4 homozygous men than women (180). Furthermore, studies on the effect of APOE-ɛ4 homozygosity in men with MCI have shown that ɛ4 men have smaller hippocampal volume than ɛ4 homozygous females (126).
4. Conclusion
Collectively, the data indicate a complex interaction between the trial of greatest risk factors for Alzheimer’s disease, age, APOE genotype and sex. The complexity of this interaction remains to be fully and specifically characterized. While investigating the interaction between three systems across multiple transition states of aging is incredibly challenging at the basic and clinical levels of analysis, it is feasible using a big data bioinformatic approach. Clearly there are sex differences in risk of AD that are modified by APOE genotype. But even at this level there is a fair degree of variability that is not explained by either APOE genotype or by sex suggesting that the systems biology of aging is driving factor.
Highlights.
The three greatest risk factors for Alzheimer’s disease are age, APOE-ɛ4 genotype and female sex.
Convergence of these three risk factors, creates unique sex differences risk profiles for Alzheimer’s disease.
The bioenergetic shift of the perimenopause to menopausal transition, unique to female, creates a risk event that likely exacerbates effect of APOE-ɛ4 positive females to thereby contribute to greater lifetime risk of Alzheimer’s disease in women.
Increased risk of earlier onset of Alzheimer’s is evident in APOE-ɛ4 homozygote males whereas greater risk of later onset AD is evident in females.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 3.Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. 2013;80:1778–1783. doi: 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayeux R, Stern Y, Ottman R, Tatemichi TK, Tang MX, Maestre G, Ngai C, Tycko B, Ginsberg H. The apolipoprotein epsilon 4 allele in patients with Alzheimer’s disease. Annals of neurology. 1993;34:752–754. doi: 10.1002/ana.410340527. [DOI] [PubMed] [Google Scholar]
- 5.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. Journal of lipid research. 2009;50(Suppl):S183–188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payami H, Montee KR, Kaye JA, Bird TD, Yu C-E, Wijsman EM, Schellenberg GD. Alzheimer’s disease, apolipoprotein E4, and gender. JAMA. 1994;271:1316–1317. [PubMed] [Google Scholar]
- 8.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. Jama. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 9.Mortensen EL, Hogh P. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology. 2001;57:89–95. doi: 10.1212/wnl.57.1.89. [DOI] [PubMed] [Google Scholar]
- 10.Damoiseaux JS, Seeley WW, Zhou J, Shirer WR, Coppola G, Karydas A, Rosen HJ, Miller BL, Kramer JH, Greicius MD. Gender modulates the APOE epsilon4 effect in healthy older adults: convergent evidence from functional brain connectivity and spinal fluid tau levels. J Neurosci. 2012;32:8254–8262. doi: 10.1523/JNEUROSCI.0305-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes LL, Wilson RS, Schneider JA, Bienias JL, Evans DA, Bennett DA. Gender, cognitive decline, and risk of AD in older persons. Neurology. 2003;60:1777–1781. doi: 10.1212/01.wnl.0000065892.67099.2a. [DOI] [PubMed] [Google Scholar]
- 12.Ruitenberg A, Ott A, van Swieten JC, Hofman A, Breteler MM. Incidence of dementia: does gender make a difference? Neurobiology of aging. 2001;22:575–580. doi: 10.1016/s0197-4580(01)00231-7. [DOI] [PubMed] [Google Scholar]
- 13.National Institute on Aging. Alzheimer’s Disease Progress Report 2014–2015: Advancing Research Toward a Cure. Bethesda: National Institute on Aging, National Institute of Health, US Department of Health and Human Services; 2015. [Google Scholar]
- 14.United Nations DoEaSAPD. World Population Ageing: 1950–2050. New York: United Nations; 2001. [Google Scholar]
- 15.United Nations DoEaSAPD. World Population Ageing 2015. 5th ed. New York: United Nations; [Google Scholar]
- 16.Masters CL, Bateman R, blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nature Reviews Disease Primers. 2015:15059. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 17.Brinton RD, Yao J, Yin F, Mack WJ, Cadenas E. Perimenopause as a neurological transition state. Nat Rev Endocrinol. 2015;11:393–405. doi: 10.1038/nrendo.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petanceska SS. Age-associated androgen deficiency and Alzheimer’s disease: a case in the making? J Alzheimers Dis. 2003;5:271–273. doi: 10.3233/jad-2003-5402. [DOI] [PubMed] [Google Scholar]
- 19.Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, Landfield PW. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blalock EM, Buechel HM, Popovic J, Geddes JW, Landfield PW. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J Chem Neuroanat. 2011;42:118–126. doi: 10.1016/j.jchemneu.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brinton RD. Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat Rev Endocrinol. 2013;9:241–250. doi: 10.1038/nrendo.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow HM, Herrup K. Genomic integrity and the ageing brain. Nat Rev Neurosci. 2015;16:672–684. doi: 10.1038/nrn4020. [DOI] [PubMed] [Google Scholar]
- 24.Curtis R, Geesaman BJ, DiStefano PS. Ageing and metabolism: drug discovery opportunities. Nat Rev Drug Discov. 2005;4:569–580. doi: 10.1038/nrd1777. [DOI] [PubMed] [Google Scholar]
- 25.Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9:57–62. doi: 10.1038/nri2471. [DOI] [PubMed] [Google Scholar]
- 26.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13:875–887. doi: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeoman M, Scutt G, Faragher R. Insights into CNS ageing from animal models of senescence. Nat Rev Neurosci. 2012;13:435–445. doi: 10.1038/nrn3230. [DOI] [PubMed] [Google Scholar]
- 28.Yin F, Yao J, Sancheti H, Feng T, Melcangi RC, Morgan TE, Finch CE, Pike CJ, Mack WJ, Cadenas E, Brinton RD. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiology of aging. 2015 doi: 10.1016/j.neurobiolaging.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klosinski LP, Yao J, Yin F, Fonteh AN, Harrington MG, Christensen TA, Trushina E, Brinton RD. White Matter Lipids as a Ketogenic Fuel Supply in Aging Female Brain: Implications for Alzheimer’s Disease. EBioMedicine. 2015;2:1888–1904. doi: 10.1016/j.ebiom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 31.Rettberg JR, Dang H, Hodis HN, Henderson VW, St John JA, Mack WJ, Brinton RD. Identifying postmenopausal women at risk for cognitive decline within a healthy cohort using a panel of clinical metabolic indicators: potential for detecting an at-Alzheimer’s risk metabolic phenotype. Neurobiology of aging. 2016;40:155–163. doi: 10.1016/j.neurobiolaging.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmans P, Hamshere M, Hollingworth P, Rice F, Tunstall N, Jones S, Moore P, Wavrant DeVrieze F, Myers A, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Williams N, Norton N, Abraham R, Kehoe P, Williams H, Rudrasingham V, O’Donovan M, Jones L, Hardy J, Goate A, Lovestone S, Owen M, Williams J. Genome screen for loci influencing age at onset and rate of decline in late onset Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2005;135B:24–32. doi: 10.1002/ajmg.b.30114. [DOI] [PubMed] [Google Scholar]
- 33.Wang JM, Irwin RW, Brinton RD. Activation of estrogen receptor alpha increases and estrogen receptor beta decreases apolipoprotein E expression in hippocampus in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16983–16988. doi: 10.1073/pnas.0608128103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leduc V, Jasmin-Belanger S, Poirier J. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol Med. 2010;16:469–477. doi: 10.1016/j.molmed.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 36.Lyall DM, Harris SE, Bastin ME, Munoz Maniega S, Murray C, Lutz MW, Saunders AM, Roses AD, Valdes Hernandez Mdel C, Royle NA, Starr JM, Porteous DJ, Wardlaw JM, Deary IJ. Alzheimer’s disease susceptibility genes APOE and TOMM40, and brain white matter integrity in the Lothian Birth Cohort 1936. Neurobiol Aging. 2014;35:1513 e1525–1533. doi: 10.1016/j.neurobiolaging.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanlon CS, Rubinsztein DC. Arginine residues at codons 112 and 158 in the apolipoprotein E gene correspond to the ancestral state in humans. Atherosclerosis. 1995;112:85–90. doi: 10.1016/0021-9150(94)05402-5. [DOI] [PubMed] [Google Scholar]
- 38.Corbo R, Scacchi R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE* 4 a ‘thrifty’allele? Annals of human genetics. 1999;63:301–310. doi: 10.1046/j.1469-1809.1999.6340301.x. [DOI] [PubMed] [Google Scholar]
- 39.Kuller LH, Shemanski L, Manolio T, Haan M, Fried L, Bryan N, Burke GL, Tracy R, Bhadelia R. Relationship between ApoE, MRI findings, and cognitive function in the Cardiovascular Health Study. Stroke. 1998;29:388–398. doi: 10.1161/01.str.29.2.388. [DOI] [PubMed] [Google Scholar]
- 40.Su YR, Blakemore JL, Zhang Y, Linton MF, Fazio S. Lentiviral Transduction of ApoAI Into Hematopoietic Progenitor Cells and Macrophages Applications to Cell Therapy of Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:1439–1446. doi: 10.1161/ATVBAHA.107.160093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyata M, Smith JD. Apolipoprotein E allele–specific antioxidant activity and effects on cytotoxicity by oxidative insults and β–amyloid peptides. Nature genetics. 1996;14:55–61. doi: 10.1038/ng0996-55. [DOI] [PubMed] [Google Scholar]
- 42.Zhong N, Weisgraber KH. Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. Journal of Biological Chemistry. 2009;284:6027–6031. doi: 10.1074/jbc.R800009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang RY, Garai K, Frieden C, Gross ML. Hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry determine the interface and dynamics of apolipoprotein E oligomerization. Biochemistry. 2011;50:9273–9282. doi: 10.1021/bi2010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Louhija J, Miettinen HE, Kontula K, Tikkanen MJ, Miettinen T, Tilvis R. Aging and genetic variation of plasma apolipoproteins. Relative loss of the apolipoprotein E4 phenotype in centenarians. Arteriosclerosis, Thrombosis, and Vascular Biology. 1994;14:1084–1089. doi: 10.1161/01.atv.14.7.1084. [DOI] [PubMed] [Google Scholar]
- 45.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annual review of genomics and human genetics. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 46.Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proceedings of the National Academy of Sciences. 1985;82:203–207. doi: 10.1073/pnas.82.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. The Journal of neuroscience. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schiele F, De Bacquer D, Vincent-Viry M, Beisiegel U, Ehnholm C, Evans A, Kafatos A, Martins M, Sans S, Sass C. Apolipoprotein E serum concentration and polymorphism in six European countries: the ApoEurope Project. Atherosclerosis. 2000;152:475–488. doi: 10.1016/s0021-9150(99)00501-8. [DOI] [PubMed] [Google Scholar]
- 49.Saunders A. Strittmatter WJ1 Schmechel D, George-Hyslop PH1 Pericak-Vance MA1 Joo SH, Rosi BL1 Gusella JF, CrapperMacLachlan DR, Alberts MJ, Hulette CM, Crain BJ, Goldgaber D, Roses AD: Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 50.Reilly S, Ferrell R, Kottke B, Sing C. The gender-specific apolipoprotein E genotype influence on the distribution of plasma lipids and apolipoproteins in the population of Rochester, Minnesota. II. Regression relationships with concomitants. American journal of human genetics. 1992;51:1311. [PMC free article] [PubMed] [Google Scholar]
- 51.Gregg RE, Zech LA, Schaefer EJ, Brewer HB. Type III hyperlipoproteinemia: defective metabolism of an abnormal apolipoprotein E. Science. 1981;211:584–586. doi: 10.1126/science.7455696. [DOI] [PubMed] [Google Scholar]
- 52.Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2010;1801:806–818. doi: 10.1016/j.bbalip.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Rasmussen KL, Tybjærg- Hansen A, Nordestgaard BG, Frikke- Schmidt R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Annals of neurology. 2015;77:301–311. doi: 10.1002/ana.24326. [DOI] [PubMed] [Google Scholar]
- 54.Gong J-S, Kobayashi M, Hayashi H, Zou K, Sawamura N, Fujita SC, Yanagisawa K, Michikawa M. Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. Journal of Biological Chemistry. 2002;277:29919–29926. doi: 10.1074/jbc.M203934200. [DOI] [PubMed] [Google Scholar]
- 55.Lane RM, Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. Journal of lipid research. 2005;46:949–968. doi: 10.1194/jlr.M400486-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Leigh-Firbank EC, Minihane AM, Leake DS, Wright JW, Murphy MC, Griffin BA, Williams CM. Eicosapentaenoic acid and docosahexaenoic acid from fish oils: differential associations with lipid responses. British Journal of Nutrition. 2002;87:435–445. doi: 10.1079/BJNBJN2002556. [DOI] [PubMed] [Google Scholar]
- 57.Ordovas JM, Schaefer EJ. Genes, variation of cholesterol and fat intake and serum lipids. Current opinion in lipidology. 1999;10:15–22. doi: 10.1097/00041433-199902000-00004. [DOI] [PubMed] [Google Scholar]
- 58.Dashti HM, Mathew TC, Hussein T, Asfar SK, Behbahani A, Khoursheed MA, Al-Sayer HM, Bo-Abbas YY, Al-Zaid NS. Long-term effects of a ketogenic diet in obese patients. Experimental & Clinical Cardiology. 2004;9:200. [PMC free article] [PubMed] [Google Scholar]
- 59.Sharman MJ, Kraemer WJ, Love DM, Avery NG, Gómez AL, Scheett TP, Volek JS. A ketogenic diet favorably affects serum biomarkers for cardiovascular disease in normal-weight men. The Journal of nutrition. 2002;132:1879–1885. doi: 10.1093/jn/132.7.1879. [DOI] [PubMed] [Google Scholar]
- 60.Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, Castellano A, Pifferi F, Bocti C, Paquet N. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition. 2011;27:3–20. doi: 10.1016/j.nut.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wood RJ, Volek JS, Liu Y, Shachter NS, Contois JH, Fernandez ML. Carbohydrate restriction alters lipoprotein metabolism by modifying VLDL, LDL, and HDL subfraction distribution and size in overweight men. The Journal of nutrition. 2006;136:384–389. doi: 10.1093/jn/136.2.384. [DOI] [PubMed] [Google Scholar]
- 62.Yancy WS, Olsen MK, Guyton JR, Bakst RP, Westman EC. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: a randomized, controlled trial. Annals of internal medicine. 2004;140:769–777. doi: 10.7326/0003-4819-140-10-200405180-00006. [DOI] [PubMed] [Google Scholar]
- 63.Mente A, de Koning L, Shannon HS, Anand SS. A systematic review of the evidence supporting a causal link between dietary factors and coronary heart disease. Archives of internal medicine. 2009;169:659–669. doi: 10.1001/archinternmed.2009.38. [DOI] [PubMed] [Google Scholar]
- 64.Lahoz C, Schaefer EJ, Cupples LA, Wilson PW, Levy D, Osgood D, Parpos S, Pedro-Botet J, Daly JA, Ordovas JM. Apolipoprotein E genotype and cardiovascular disease in the Framingham Heart Study. Atherosclerosis. 2001;154:529–537. doi: 10.1016/s0021-9150(00)00570-0. [DOI] [PubMed] [Google Scholar]
- 65.Wallenfeldt K, Bokemark L, Wikstrand J, Hulthe J, Fagerberg B. Apolipoprotein B/apolipoprotein AI in relation to the metabolic syndrome and change in carotid artery intima-media thickness during 3 years in middle-aged men. Stroke. 2004;35:2248–2252. doi: 10.1161/01.STR.0000140629.65145.3c. [DOI] [PubMed] [Google Scholar]
- 66.Shalaurova I, Connelly MA, Garvey WT, Otvos JD. Lipoprotein Insulin Resistance Index: A Lipoprotein Particle–Derived Measure of Insulin Resistance. Metabolic syndrome and related disorders. 2014;12:422–429. doi: 10.1089/met.2014.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olano-Martin E, Anil E, Caslake MJ, Packard CJ, Bedford D, Stewart G, Peiris D, Williams CM, Minihane AM. Contribution of apolipoprotein E genotype and docosahexaenoic acid to the LDL-cholesterol response to fish oil. Atherosclerosis. 2010;209:104–110. doi: 10.1016/j.atherosclerosis.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 68.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proceedings of the National Academy of Sciences. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cramer PE, Cirrito JR, Wesson DW, Lee CD, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ. ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoang MH, Jia Y, Jun HJ, Lee JH, Lee BY, Lee SJ. Fucosterol is a selective liver X receptor modulator that regulates the expression of key genes in cholesterol homeostasis in macrophages, hepatocytes, and intestinal cells. Journal of agricultural and food chemistry. 2012;60(46):11567–11575. doi: 10.1021/jf3019084. [DOI] [PubMed] [Google Scholar]
- 72.Roses A, Lutz M, Amrine-Madsen H, Saunders A, Crenshaw D, Sundseth S, Huentelman M, Welsh-Bohmer K, Reiman E. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. The pharmacogenomics journal. 2010;10:375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soares H, Potter W, Pickering E, Kuhn M, Immermann F, Shera D, Ferm M, Dean R, Simon A, Swenson F. Biomarkers Consortium Alzheimer’s Disease Plasma Proteomics Project. Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Arch Neurol. 2012;69:1310–1317. doi: 10.1001/archneurol.2012.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shachter N, Hayek T, Leff T, Smith J, Rosenberg D, Walsh A, Ramakrishnan R, Goldberg I, Ginsberg H, Breslow J. Overexpression of apolipoprotein CII causes hypertriglyceridemia in transgenic mice. Journal of Clinical Investigation. 1994;93:1683. doi: 10.1172/JCI117151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Middelberg RP, Ferreira MA, Henders AK, Heath AC, Madden PA, Montgomery GW, Martin NG, Whitfield JB. Genetic variants in LPL, OASL and TOMM40/APOE-C1-C2-C4 genes are associated with multiple cardiovascular-related traits. BMC medical genetics. 2011;12:123. doi: 10.1186/1471-2350-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Medeiros LA, Khan T, El Khoury JB, Pham CL, Hatters DM, Howlett GJ, Lopez R, O’Brien KD, Moore KJ. Fibrillar amyloid protein present in atheroma activates CD36 signal transduction. Journal of Biological Chemistry. 2004;279:10643–10648. doi: 10.1074/jbc.M311735200. [DOI] [PubMed] [Google Scholar]
- 77.Serra-Grabulosa J, Salgado-Pineda P, Junqué C, Sole-Padulles C, Moral P, López-Alomar A, Lopez T, Lopez-Guillen A, Bargallo N, Mercader J. Apolipoproteins E and C1 and brain morphology in memory impaired elders. Neurogenetics. 2003;4:141–146. doi: 10.1007/s10048-002-0142-8. [DOI] [PubMed] [Google Scholar]
- 78.Garai K, Baban B, Frieden C. Dissociation of apolipoprotein E oligomers to monomer is required for high-affinity binding to phospholipid vesicles. Biochemistry. 2011;50:2550–2558. doi: 10.1021/bi1020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.LaDu MJ, Munson GW, Jungbauer L, Getz GS, Reardon CA, Tai LM, Yu C. Preferential interactions between ApoE-containing lipoproteins and Abeta revealed by a detection method that combines size exclusion chromatography with non-reducing gel-shift. Biochim Biophys Acta. 2012;1821:295–302. doi: 10.1016/j.bbalip.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manelli AM, Stine WB, Van Eldik LJ, LaDu MJ. ApoE and Abeta1-42 interactions: effects of isoform and conformation on structure and function. J Mol Neurosci. 2004;23:235–246. doi: 10.1385/JMN:23:3:235. [DOI] [PubMed] [Google Scholar]
- 81.Tai LM, Mehra S, Shete V, Estus S, Rebeck GW, Bu G, LaDu MJ. Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegener. 2014;9(2) doi: 10.1186/1750-1326-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. American Journal of Physiology-Cell Physiology. 2007;292:C1256–C1262. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- 84.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, Ackermann U, Jones G, Szoeke C, Salvado O. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Annals of neurology. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Apostolova LG, Hwang KS, Kohannim O, Avila D, Elashoff D, Jack CR, Shaw L, Trojanowski JQ, Weiner MW, Thompson PM. ApoE4 effects on automated diagnostic classifiers for mild cognitive impairment and Alzheimer’s disease. NeuroImage: Clinical. 2014;4:461–472. doi: 10.1016/j.nicl.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bennett RE, Esparza TJ, Lewis HA, Kim E, Mac Donald CL, Sullivan PM, Brody DL. Human apolipoprotein E4 worsens acute axonal pathology but not amyloid-β immunoreactivity after traumatic brain injury in 3xTG-AD mice. Journal of Neuropathology & Experimental Neurology. 2013;72:396–403. doi: 10.1097/NEN.0b013e31828e24ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Strittmatter W, Weisgraber K, Goedert M, Saunders A, Huang D, Corder E, Dong L, Jakes R, Alberts M, Gilbert J. Hypothesis: microtubule instability and paired. 1994 doi: 10.1006/exnr.1994.1019. [DOI] [PubMed] [Google Scholar]
- 89.Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka S-K, van der Flier WM, Blankenstein MA, Ewers M. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. Jama. 2009;302:385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 90.Ohno-Iwashita Y, Shimada Y, Waheed AA, Hayashi M, Inomata M, Nakamura M, Maruya M, Iwashita S. Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe. 2004;10:125–134. doi: 10.1016/j.anaerobe.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 91.Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proceedings of the National Academy of Sciences. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brecht WJ, Harris FM, Chang S, Tesseur I, Yu G-Q, Xu Q, Fish JD, Wyss-Coray T, Buttini M, Mucke L. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. The Journal of neuroscience. 2004;24:2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Conejero-Goldberg C, Hyde T, Chen S, Dreses-Werringloer U, Herman M, Kleinman J, Davies P, Goldberg T. Molecular signatures in post-mortem brain tissue of younger individuals at high risk for Alzheimer’s disease as based on APOE genotype. Molecular psychiatry. 2011;16:836–847. doi: 10.1038/mp.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crivello F, Lemaître H, Dufouil C, Grassiot B, Delcroix N, Tzourio-Mazoyer N, Tzourio C, Mazoyer B. Effects of ApoE-ɛ4 allele load and age on the rates of grey matter and hippocampal volumes loss in a longitudinal cohort of 1186 healthy elderly persons. Neuroimage. 2010;53:1064–1069. doi: 10.1016/j.neuroimage.2009.12.116. [DOI] [PubMed] [Google Scholar]
- 96.Ihara Y, Hayabara T, Sasaki K, Kawada R, Nakashima Y, Kuroda S. Relationship between oxidative stress and apoE phenotype in Alzheimer’s disease. Acta neurologica scandinavica. 2000;102:346–349. doi: 10.1034/j.1600-0404.2000.102006346.x. [DOI] [PubMed] [Google Scholar]
- 97.Gau B, Garai K, Frieden C, Gross ML. Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry. 2011;50:8117–8126. doi: 10.1021/bi200911c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramassamy C, Averill D, Beffert U, Bastianetto S, Theroux L, Lussier-Cacan S, Cohn JS, Christen Y, Davignon J, Quirion R. Oxidative damage and protection by antioxidants in the frontal cortex of Alzheimer’s disease is related to the apolipoprotein E genotype. Free Radical Biology and Medicine. 1999;27:544–553. doi: 10.1016/s0891-5849(99)00102-1. [DOI] [PubMed] [Google Scholar]
- 99.Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, Freedman SB, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. The Journal of biological chemistry. 2011;286:5215–5221. doi: 10.1074/jbc.M110.151084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jarvik G, Wijsman EM, Kukull W, Schellenberg G, Yu C, Larson E. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer’s disease A case-control study. Neurology. 1995;45:1092–1096. doi: 10.1212/wnl.45.6.1092. [DOI] [PubMed] [Google Scholar]
- 101.Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiology of aging. 2004;25:641–650. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 102.Noguchi S, Murakami K, Yamada N, Payami H, Kaye J, Heston L, Bird T, Schellenberg G. Apolipoprotein E genotype and Alzheimer’s disease. The Lancet. 1993;342:737–738. [PubMed] [Google Scholar]
- 103.Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, Zismann VL, Beach TG, Leung D, Bryden L. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. Journal of Clinical Psychiatry. 2007;68:613. doi: 10.4088/jcp.v68n0419. [DOI] [PubMed] [Google Scholar]
- 104.Corder E, Saunders A, Strittmatter W, Schmechel D, Gaskell P, Small G, Roses A, Haines J, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 105.Mondadori CR, Dominique J-F, Buchmann A, Mustovic H, Wollmer MA, Schmidt CF, Boesiger P, Hock C, Nitsch RM, Papassotiropoulos A. Better memory and neural efficiency in young apolipoprotein E ɛ4 carriers. Cerebral Cortex. 2007;17:1934–1947. doi: 10.1093/cercor/bhl103. [DOI] [PubMed] [Google Scholar]
- 106.Marchant NL, King SL, Tabet N, Rusted JM. Positive effects of cholinergic stimulation favor young APOE ɛ 4 carriers. Neuropsychopharmacology. 2010;35:1090–1096. doi: 10.1038/npp.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Evans S, Dowell NG, Tabet N, Tofts PS, King SL, Rusted JM. Cognitive and neural signatures of the APOE E4 allele in mid-aged adults. Neurobiology of aging. 2014;35:1615–1623. doi: 10.1016/j.neurobiolaging.2014.01.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer’s disease. New England journal of medicine. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smith C, Andersen A, Kryscio R, Schmitt F, Kindy M, Blonder L, Avison M. Women at risk for AD show increased parietal activation during a fluency task. Neurology. 2002;58:1197–1202. doi: 10.1212/wnl.58.8.1197. [DOI] [PubMed] [Google Scholar]
- 110.Sheffler J, Moxley J, Sachs-Ericsson N. Stress, race, and APOE: understanding the interplay of risk factors for changes in cognitive functioning. Aging & mental health. 2014;18:784–791. doi: 10.1080/13607863.2014.880403. [DOI] [PubMed] [Google Scholar]
- 111.Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, Clasen L, Evans A, Rapoport JL, Giedd JN. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. The Lancet Neurology. 2007;6:494–500. doi: 10.1016/S1474-4422(07)70106-0. [DOI] [PubMed] [Google Scholar]
- 112.Moffat S, Szekely C, Zonderman A, Kabani N, Resnick S. Longitudinal change in hippocampal volume as a function of apolipoprotein E genotype. Neurology. 2000;55:134–136. doi: 10.1212/wnl.55.1.134. [DOI] [PubMed] [Google Scholar]
- 113.Bartzokis G, Lu PH, Geschwind DH, Tingus K, Huang D, Mendez MF, Edwards N, Mintz J. Apolipoprotein E affects both myelin breakdown and cognition: implications for age-related trajectories of decline into dementia. Biological psychiatry. 2007;62:1380–1387. doi: 10.1016/j.biopsych.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 114.Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D’Angelo G, Liu C, Dixit S, Benzinger T, Fagan A. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. The Journal of Neuroscience. 2010;30:17035–17040. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proceedings of the National Academy of Sciences. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E ɛ4 gene dose and brain-imaging measurements of regional hypometabolism. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8299–8302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mosconi L, Perani D, Sorbi S, Herholz K, Nacmias B, Holthoff V, Salmon E, Baron JC, De Cristofaro MT, Padovani A, Borroni B, Franceschi M, Bracco L, Pupi A. MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology. 2004;63:2332–2340. doi: 10.1212/01.wnl.0000147469.18313.3b. [DOI] [PubMed] [Google Scholar]
- 118.Chiang G, Zhan W, Schuff N, Weiner M. White Matter Alterations in Cognitively Normal apoE ɛ2 Carriers: Insight into Alzheimer Resistance? American Journal of Neuroradiology. 2012;33:1392–1397. doi: 10.3174/ajnr.A2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alzheimer’s Disease I. World Alzheimer Report 2015 The Global Impact of Dementia: An analysis of prevalence, incidence, cost and trends. London: 2015. [Google Scholar]
- 121.Altmann A, Tian L, Henderson VW, Greicius MD. Sex modifies the APOE-related risk of developing Alzheimer disease. Annals of neurology. 2014;75:563–573. doi: 10.1002/ana.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Archives of general psychiatry. 2005;62:685–691. doi: 10.1001/archpsyc.62.6.685. [DOI] [PubMed] [Google Scholar]
- 123.Lin KA, Choudhury KR, Rathakrishnan BG, Marks DM, Petrella JR, Doraiswamy PM. Marked gender differences in progression of mild cognitive impairment over 8 years. Alzheimers Dement (N Y) 2015;1:103–110. doi: 10.1016/j.trci.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Corder E, Saunders A, Strittmatter W, Schmechel D, Gaskell P, Rimmler J, Locke P, Conneally P, Schmader K, Tanzi R. Apolipoprotein E, survival in Alzheimer’s disease patients, and the competing risks of death and Alzheimer’s disease. Neurology. 1995;45:1323–1328. doi: 10.1212/wnl.45.7.1323. [DOI] [PubMed] [Google Scholar]
- 125.Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the ɛ4 allele for apolipoprotein E. New England Journal of Medicine. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 126.Fleisher A, Grundman M, Jack CR, Petersen RC, Taylor C, Kim HT, Schiller DH, Bagwell V, Sencakova D, Weiner MF. Sex, apolipoprotein E ɛ4 status, and hippocampal volume in mild cognitive impairment. Archives of neurology. 2005;62:953–957. doi: 10.1001/archneur.62.6.953. [DOI] [PubMed] [Google Scholar]
- 127.Jack CR, Wiste HJ, Weigand SD, Knopman DS, Vemuri P, Mielke MM, Lowe V, Senjem ML, Gunter JL, Machulda MM. Age, Sex, and APOE ɛ4 Effects on Memory, Brain Structure, and β-Amyloid Across the Adult Life Span. JAMA neurology. 2015;72:511–519. doi: 10.1001/jamaneurol.2014.4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Corder EH, Ghebremedhin E, Taylor MG, Thal DR, OHM TG, Braak H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Annals of the New York Academy of Sciences. 2004;1019:24–28. doi: 10.1196/annals.1297.005. [DOI] [PubMed] [Google Scholar]
- 129.Yaffe K, Haan M, Byers A, Tangen C, Kuller L. Estrogen use, APOE, and cognitive decline: evidence of gene-environment interaction. Neurology. 2000;54:1949–1954. doi: 10.1212/wnl.54.10.1949. [DOI] [PubMed] [Google Scholar]
- 130.Newhouse P, Albert K, Astur R, Johnson J, Naylor M, Dumas J. Tamoxifen improves cholinergically modulated cognitive performance in postmenopausal women. Neuropsychopharmacology. 2013;38:2632–2643. doi: 10.1038/npp.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ding F, Yao J, Rettberg JR, Chen S, Brinton RD. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PloS one. 2013;8:e79977. doi: 10.1371/journal.pone.0079977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rettberg JR, Yao J, Brinton RD. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol. 2014;35:8–30. doi: 10.1016/j.yfrne.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ding F, Yao J, Zhao L, Mao Z, Chen S, Brinton RD. Ovariectomy induces a shift in fuel availability and metabolism in the hippocampus of the female transgenic model of familial Alzheimer’s. PloS one. 2013;8:e59825. doi: 10.1371/journal.pone.0059825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yao J, Brinton RD. Estrogen regulation of mitochondrial bioenergetics: implications for prevention of Alzheimer’s disease. Adv Pharmacol. 2012;64:327–371. doi: 10.1016/B978-0-12-394816-8.00010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yao J, Rettberg JR, Klosinski LP, Cadenas E, Brinton RD. Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol Aspects Med. 2011;32:247–257. doi: 10.1016/j.mam.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yao J, Irwin R, Chen S, Hamilton R, Cadenas E, Brinton RD. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta-amyloid. Neurobiology of aging. 2012;33:1507–1521. doi: 10.1016/j.neurobiolaging.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yao J, Hamilton RT, Cadenas E, Brinton RD. Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim Biophys Acta. 2010;1800:1121–1126. doi: 10.1016/j.bbagen.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Langbaum JB, Chen K, Caselli RJ, Lee W, Reschke C, Bandy D, Alexander GE, Burns CM, Kaszniak AW, Reeder SA, Corneveaux JJ, Allen AN, Pruzin J, Huentelman MJ, Fleisher AS, Reiman EM. Hypometabolism in Alzheimer-affected brain regions in cognitively healthy Latino individuals carrying the apolipoprotein E epsilon4 allele. Archives of neurology. 2010;67:462–468. doi: 10.1001/archneurol.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci U S A. 2005;102:8299–8302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ryan J, Carriere I, Carcaillon L, Dartigues J-F, Auriacombe S, Rouaud O, Berr C, Ritchie K, Scarabin P-Y, Ancelin M-L. Estrogen receptor polymorphisms and incident dementia: The prospective 3C study. Alzheimer’s & Dementia. 2014;10:27–35. doi: 10.1016/j.jalz.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 142.Darabi M, Ani M, Panjehpour M, Rabbani M, Movahedian A, Zarean E. Effect of estrogen receptor β A1730G polymorphism on ABCA1 gene expression response to postmenopausal hormone replacement therapy. Genetic testing and molecular biomarkers. 2011;15:11–15. doi: 10.1089/gtmb.2010.0106. [DOI] [PubMed] [Google Scholar]
- 143.Srivastava RAK. Estrogen-induced regulation of the ATP-binding cassette transporter A1 (ABCA1) in mice: a possible mechanism of atheroprotection by estrogen. Molecular and cellular biochemistry. 2002;240:67–73. doi: 10.1023/a:1020604610873. [DOI] [PubMed] [Google Scholar]
- 144.Jacobs EG, Kroenke C, Lin J, Epel ES, Kenna HA, Blackburn EH, Rasgon NL. Accelerated cell aging in female APOE-epsilon4 carriers: implications for hormone therapy use. PloS one. 2013;8:e54713. doi: 10.1371/journal.pone.0054713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pedram A, Razandi M, Wallace DC, Levin ER. Functional estrogen receptors in the mitochondria of breast cancer cells. Molecular biology of the cell. 2006;17:2125–2137. doi: 10.1091/mbc.E05-11-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gabriel K, Egan B, Lithgow T. Tom40, the import channel of the mitochondrial outer membrane, plays an active role in sorting imported proteins. The EMBO journal. 2003;22:2380–2386. doi: 10.1093/emboj/cdg229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Srivastava RAK, Srivastava N, Averna M, Lin RC, Korach KS, Lubahn DB, Schonfeld G. Estrogen up-regulates apolipoprotein E (ApoE) gene expression by increasing ApoE mRNA in the translating pool via the estrogen receptor α-mediated pathway. Journal of Biological Chemistry. 1997;272:33360–33366. doi: 10.1074/jbc.272.52.33360. [DOI] [PubMed] [Google Scholar]
- 148.Cheng X, McAsey ME, Li M, Randall S, Cady C, Nathan BP, Struble RG. Estradiol replacement increases the low-density lipoprotein receptor related protein (LRP) in the mouse brain. Neuroscience letters. 2007;417:50–54. doi: 10.1016/j.neulet.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 149.Ivanova MM, Radde BN, Son J, Mehta FF, Chung S-H, Klinge CM. Estradiol and tamoxifen regulate NRF-1 and mitochondrial function in mouse mammary gland and uterus. Journal of molecular endocrinology. 2013;51:233–246. doi: 10.1530/JME-13-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Struble RG, Cady C, Nathan BP, McAsey M. Apolipoprotein E may be a critical factor in hormone therapy neuroprotection. Frontiers in bioscience: a journal and virtual library. 2007;13:5387–5405. doi: 10.2741/3088. [DOI] [PubMed] [Google Scholar]
- 151.Nathan BP, Barsukova AG, Shen F, McAsey M, Struble RG. Estrogen facilitates neurite extension via apolipoprotein E in cultured adult mouse cortical neurons. Endocrinology. 2004;145:3065–3073. doi: 10.1210/en.2003-1707. [DOI] [PubMed] [Google Scholar]
- 152.Dean DC, Jerskey BA, Chen K, Protas H, Thiyyagura P, Roontiva A, O’Muircheartaigh J, Dirks H, Waskiewicz N, Lehman K. Brain differences in infants at differential genetic risk for late-onset Alzheimer disease: a cross-sectional imaging study. JAMA neurology. 2014;71:11–22. doi: 10.1001/jamaneurol.2013.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bartzokis G, Lu PH, Geschwind DH, Edwards N, Mintz J, Cummings JL. Apolipoprotein E genotype and age-related myelin breakdown in healthy individuals: implications for cognitive decline and dementia. Archives of General Psychiatry. 2006;63:63–72. doi: 10.1001/archpsyc.63.1.63. [DOI] [PubMed] [Google Scholar]
- 154.Vincent-Viry M, Schiele F, Gueguen R, Bohnet K, Visvikis S, Siest G. Biological variations and genetic reference values for apolipoprotein E serum concentrations: results from the STANISLAS cohort study. Clinical chemistry. 1998;44:957–965. [PubMed] [Google Scholar]
- 155.LEE PA, GUO SS, KULIN HE. Age of puberty: data from the United States of America. Apmis. 2001;109:S156–S163. doi: 10.1034/j.1600-0463.2001.d01-107.x. [DOI] [PubMed] [Google Scholar]
- 156.Burger HG, Dudley EC, Robertson DM, Dennerstein L. Hormonal changes in the menopause transition. Recent Progress in Hormone Research. 2002;57:257–276. doi: 10.1210/rp.57.1.257. [DOI] [PubMed] [Google Scholar]
- 157.Yao J, Hamilton RT, Cadenas E, Brinton RD. Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochimica et Biophysica Acta (BBA)-General Subjects. 2010;1800:1121–1126. doi: 10.1016/j.bbagen.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yao J, Irwin R, Chen S, Hamilton R, Cadenas E, Brinton RD. Ovarian hormone loss induces bioenergetic deficits and mitochondrial β-amyloid. Neurobiology of aging. 2012;33:1507–1521. doi: 10.1016/j.neurobiolaging.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li R, Cui J, Shen Y. Brain sex matters: estrogen in cognition and Alzheimer’s disease. Molecular and cellular endocrinology. 2014;389:13–21. doi: 10.1016/j.mce.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Santoro N, Sutton-Tyrrell K. The SWAN song: Study of Women’s Health Across the Nation’s recurring themes. Obstetrics and gynecology clinics of North America. 2011;38:417–423. doi: 10.1016/j.ogc.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Garatachea N, Emanuele E, Calero M, Fuku N, Arai Y, Abe Y, Murakami H, Miyachi M, Yvert T, Verde Z. ApoE gene and exceptional longevity: insights from three independent cohorts. Experimental gerontology. 2014;53:16–23. doi: 10.1016/j.exger.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 162.Asada T, Kariya T, Yamagata Z, Kinoshita T, Asaka A. Apolipoprotein E allele in centenarians. Neurology. 1996;46:1484–1484. doi: 10.1212/wnl.46.5.1484. [DOI] [PubMed] [Google Scholar]
- 163.Blanché H, Cabanne L, Sahbatou M, Thomas G. A study of French centenarians: are ACE and APOE associated with longevity? Comptes Rendus de l’Académie des Sciences-Series III-Sciences de la Vie. 2001;324:129–135. doi: 10.1016/s0764-4469(00)01274-9. [DOI] [PubMed] [Google Scholar]
- 164.Castro E, Ogburn CE, Hunt KE, Tilvis R, Louhija J, Penttinen R, Erkkola R, Panduro A, Riestra R, Piussan C. Polymorphisms at the Werner locus: I. Newly identified polymorphisms, ethnic variability of 1367Cy/Arg, and its stability in a population of Finnish centenarians. American journal of medical genetics. 1999;82:399–403. [PubMed] [Google Scholar]
- 165.Schachter F, Faure-Delanef L, Guénot F, Rouger H, Froguel P, Lesueur-Ginot L, Cohen D. Genetic associations with human longevity at the APOE and ACE loci. Nature genetics. 1994;6:29–32. doi: 10.1038/ng0194-29. [DOI] [PubMed] [Google Scholar]
- 166.Qiu C, Kivipelto M, Agüero-Torres H, Winblad B, Fratiglioni L. Risk and protective effects of the APOE gene towards Alzheimer’s disease in the Kungsholmen project: variation by age and sex. Journal of Neurology, Neurosurgery & Psychiatry. 2004;75:828–833. doi: 10.1136/jnnp.2003.021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rosvall L, Rizzuto D, Wang HX, Winblad B, Graff C, Fratiglioni L. APOE-related mortality: effect of dementia, cardiovascular disease and gender. Neurobiology of aging. 2009;30:1545–1551. doi: 10.1016/j.neurobiolaging.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 168.Kuhel DG, Konaniah ES, Basford JE, McVey C, Goodin CT, Chatterjee TK, Weintraub NL, Hui DY. Apolipoprotein E2 accentuates postprandial inflammation and diet-induced obesity to promote hyperinsulinemia in mice. Diabetes. 2013;62:382–391. doi: 10.2337/db12-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Satirapoj B, Supasyndh O, Dispan R, Punpanich D, Tribanyatkul S, Choovichian P. Apolipoprotein E genetic polymorphisms and the development of nephropathy in type 2 diabetes. J Med Assoc Thai. 2013;96:1119–1126. [PubMed] [Google Scholar]
- 170.Nebel A, Kleindorp R, Caliebe A, Nothnagel M, Blanché H, Junge O, Wittig M, Ellinghaus D, Flachsbart F, Wichmann H-E. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mechanisms of ageing and development. 2011;132:324–330. doi: 10.1016/j.mad.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 171.Galinsky D, Tysoe C, Brayne CE, Easton DF, Huppert FA, Dening TR, Paykel ES, Rubinsztein DC. Analysis of the apo E/apo CI, angiotensin converting enzyme and methylenetetrahydrofolate reductase genes as candidates affecting human longevity. Atherosclerosis. 1997;129:177–183. doi: 10.1016/s0021-9150(96)06027-3. [DOI] [PubMed] [Google Scholar]
- 172.Morris JC, Weintraub S, Chui HC, Cummings J, DeCarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Disease & Associated Disorders. 2006;20:210–216. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- 173.Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, Hubbard JL, Koepsell TD, Morris JC, Kukull WA. The National Alzheimer’s Coordinating Center (NACC) database: the uniform data set. Alzheimer Disease & Associated Disorders. 2007;21:249–258. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 174.Chêne G, Beiser A, Au R, Preis SR, Wolf PA, Dufouil C, Seshadri S. Gender and incidence of dementia in the Framingham Heart Study from mid-adult life. Alzheimer’s & Dementia. 2015;11:310–320. doi: 10.1016/j.jalz.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.De Marinis E, Martini C, Trentalance A, Pallottini V. Sex differences in hepatic regulation of cholesterol homeostasis. Journal of Endocrinology. 2008;198:635–643. doi: 10.1677/JOE-08-0242. [DOI] [PubMed] [Google Scholar]
- 176.Brussaard H, Leuven JG, Frölich M, Kluft C, Krans H. Short-term oestrogen replacement therapy improves insulin resistance, lipids and fibrinolysis in postmenopausal women with NIDDM. Diabetologia. 1997;40:843–849. doi: 10.1007/s001250050758. [DOI] [PubMed] [Google Scholar]
- 177.Miech R, Breitner J, Zandi P, Khachaturian A, Anthony J, Mayer L. Incidence of AD may decline in the early 90s for men, later for women The Cache County study. Neurology. 2002;58:209–218. doi: 10.1212/wnl.58.2.209. [DOI] [PubMed] [Google Scholar]
- 178.Kaerst L, Kuhlmann A, Wedekind D, Stoeck K, Lange P, Zerr I. Cerebrospinal fluid biomarkers in Alzheimer’s disease, vascular dementia and ischemic stroke patients: a critical analysis. Journal of neurology. 2013;260:2722–2727. doi: 10.1007/s00415-013-7047-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Teng E, Chow N, Hwang KS, Thompson PM, Gylys KH, Cole GM, Jack CR, Jr, Shaw LM, Trojanowski JQ, Soares HD. Low Plasma ApoE levels are associated with smaller hippocampal size in the Alzheimer’s disease neuroimaging initiative cohort. Dementia and geriatric cognitive disorders. 2015;39:154–166. doi: 10.1159/000368982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lehmann D, Refsum H, Nurk E, Warden D, Tell G, Vollset S, Engedal K, Nygaard H, Smith A. Apolipoprotein E ɛ4 and impaired episodic memory in community-dwelling elderly people: a marked sex difference. The Hordaland Health Study. Journal of Neurology, Neurosurgery & Psychiatry. 2006;77:902–908. doi: 10.1136/jnnp.2005.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]