

Abstract
B-cell activating factor of the TNF family (BAFF) has been documented to act as a critical factor in the development of aggressive B lymphocytes and autoimmune diseases. However, the effect of various cytokines on BAFF-elicited neoplastic B-lymphoid cells is not known. In this study, we exhibited that administration of human soluble BAFF (hsBAFF), IL-2, IL-4, IFN-γ, or TNF-α alone increased cell viability and survival in Raji cells concentration-dependently, yet a more robust viability/survival was seen in the cells co-treatment of IL-2, IL-4, IFN-γ, or TNF-α with hsBAFF, respectively. Further research revealed that both Erk1/2 and S6K1 signaling pathways were essential for IL-2, IL-4, IFN-γ, or TNF-α enhancement of the viability/survival in the hsBAFF-stimulated cells, as inhibition of Erk1/2 with U0126 or down-regulation of Erk1/2, or blockage of S6K1 with rapamycin or silencing S6K1, or silencing S6K1/Erk1/2, respectively, reduced the cell viability/survival in the cells treated with/without hsBAFF ± IL-2, IL-4, IFN-γ, or TNF-α. These findings indicate that IL-2, IL-4, IFN-γ or TNF-α enhances BAFF-stimulated cell viability/survival by activating Erk1/2 and S6K1 signaling in neoplastic B-lymphoid cells. Our data suggest that modulation of IL-2, IL-4, IFN-γ and/or TNF-α levels, or inhibitors of Erk1/2 or S6K1 may be a new approach to prevent BAFF-induced aggressive B-cell malignancies.
Keywords: Cytokine, BAFF, B cell, Erk1/2, S6K1
1. Introduction
B-cell activating factor of the TNF family (BAFF), a type II membrane protein that exists in both membrane-bound and soluble form, is also known as BLyS, TALL-1, THANK, and zTNF4 [1–4]. BAFF may be expressed by monocytes, activated neutrophils, T cells and dendritic cells and act as a ligand through three TNF-receptor-family members: BAFF-R (BR3), BCMA and TACI, and [1–10]. Evidence shows that BAFF is an important regulator of B-cell development, differentiation, maturation, proliferation and survival, as well as immune responses [11–14]. Lack of BAFF causes B lymphocyte reduction and dysfunction of immune system, with symptoms including single IgA, IgG, or IgM deficiency and common variable immunodeficiency [15, 16]. Conversely, high levels of BAFF promote cell proliferation/survival of peripheral B lymphocytes, and thus leading to continued production of plasma cells releasing pathogenic autoantibodies, which is implicated in various autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and Sjögren’s syndrome (SS) [17–21]. These results highlight that BAFF is critical for the development of normal B cells as well as aggressive and malignant B cells. Especially, the malignant B cells with prolonged lifespan may be culprits in human B-cell malignancies related to autoimmune diseases.
In addition to BAFF, some other cytokines also affect B-cell proliferation/survival. For example, interleukin (IL)-2 evokes proliferation of activated B cells through surface receptors and induces human plasma cell differentiation [22, 23]. IL-4, as a vital growth and survival factor for normal B cells, promotes B cell proliferation/viability and confers resistance to apoptosis [24]. Interferon (IFN)-γ enhances the secretion of immunoglobulin (Ig) in activated murine and human B cells, and promotes the entrance to S phase of cell cycle in human B cells [25]. TNF-α may play a costimulatory role in mitogen-activated normal human B cells and act as a possible signal for proliferative of chronic B-leukemia cells, suggesting a potential regulatory effect of TNF-α on normal and neoplastic B-lymphoid cells [26–30]. Taken together, these data indicate that IL-2, IL-4, IFN-γ and TNF-α exert a key role in the modulation of cell proliferation/survival and Ig secretion in normal and neoplastic B-lymphoid cells [22, 23, 25–30]. However, whether and how such cytokines affect BAFF-promoted B-cell proliferation/survival is unknown.
The extracellular signal-related kinases 1/2 (Erk1/2), a member of mitogen-activated protein kinases (MAPKs) family, plays a vital action in the proliferation and survival of different types of cells [31, 32]. The mammalian/mechanistic target of rapamycin (mTOR), a central controller for regulating cell proliferation/survival [33, 34], lies downstream of phosphatidylinositol 3′-kinase (PI3K) and protein kinase B (PKB/Akt) [35, 36]. Activated PI3K/Akt may positively regulate mTOR, which further causes phosphorylation of ribosomal p70 S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1), the two best-characterized downstream effector molecules of mTOR [34, 37]. Many data have shown IL-2, IL-4, IFN-γ, or TNF-α can activate Erk1/2 and PI3K/Akt/S6K1 pathways [23, 38, 39]. Recently, our group has unveiled that human soluble BAFF (hsBAFF) promotes cell proliferation/survival not only in an Erk1/2-dependent nmanner, but also partially via activation of mTOR-mediated S6K1 and 4E-BP1 pathways in normal and neoplastic B-lymphoid cells [40–42]. This prompted us to study the potential role of Erk1/2 and S6K1 activity in IL-2/IL-4/IFN-γ/TNF-α’s effect on hsBAFF-stimulated B-cell viability/survival.
Here we show that hsBAFF-stimulated cell viability/survival was significantly strengthened by co-treatment with IL-2, IL-4, IFN-γ, or TNF-α in Raji cells. We identified that both Erk1/2 and S6K1 signaling pathways were required for IL-2, IL-4, IFN-γ, or TNF-α enhancement of cell viability/survival in hsBAFF-stimulated Raji cells. The results indicate that these cytokines enhance BAFF-stimulated cell viability/survival by activating Erk1/2 and S6K1 pathways in neoplastic B-lymphoid cells. Our findings underscore that manipulation of IL-2, IL-4, IFN-γ and/or TNF-α levels, or inhibitors of Erk1/2 or S6K1 may be a new approach to prevent BAFF-induced aggressive B-cell malignancies.
2. Materials and methods
2.1. Reagents
IL-2, IL-4, IFN-γ, and TNF-α were supplied by Peprotech (Rocky Hill, NJ, USA). Refolded hsBAFF was a recombinant form of the extracellular domain of the BAFF synthesized in Escherichia coli from our group [43]. Rapamycin was purchased from ALEXIS (San Diego, CA, USA), whereas U0126 was from Sigma (St. Louis, MO, USA). RPMI 1640 Medium was from Gibco (Rockville, MD, USA). Fetal bovine serum (FBS) was purchased from Hyclone (Logan, UT, USA). CellTiter 96® AQueous One Solution Cell Proliferation Assay kit was provided by Promega (Madison, WI, USA). Annexin V-FITC/propidium iodide (PI) Apoptosis Detection kit was from BD Biosciences (San Diego, CA, USA). Enhanced chemiluminescence solution was from Millipore (Billerica, MA, USA). Other chemicals used in this work are of analytical grade and were obtained from Sigma and local commercial sources.
2.2. Cell culture
Neoplastic B-lymphoid (Raji) cell line (American Type Culture Collection, Manassas, VA, USA) was cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin in a humidified incubator of 5% CO2 at 37°C.
2.3. Lentiviral shRNA cloning and infection of cells
Lentiviral shRNA to Erk1/2, S6K1, S6K1/Erk1/2 and green fluorescence protein (GFP) (for control) were constructed and infected as described previously [44, 45].
2.4. MTS assay for cell viability and live cell counting by trypan blue exclusion
Raji cells, or Raji cells infected with lentiviral shRNAs to S6K1, Erk1/2 and GFP, respectively, were seeded in 96-well plates (3×104 cells/well, for cell viability assay) or 24-well plates (3×105 cells/well, for trypan blue exclusion) and cultured for overnight in humidified incubator of 5% CO2 at 37°C. Next day, cells were treated with hsBAFF (0–0.25 μg/ml), IL-2 (0–100 ng/ml), IL-4 (0–100 ng/ml), IFN-γ (0–100 ng/ml) or TNF-α (0–100 ng/ml) for 48 h, or treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (5 and/or 50 ng/ml), IL-4 (5 and/or 25 ng/ml), IFN-γ (10 and/or 100 ng/ml) or TNF-α (5 and/or 50 ng/ml) for 48 h, or pre-incubated with/without U0126 (5 μM) for 1 h or rapamycin (0.2 μg/ml) for 2 h and then treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (50 ng/ml), IL-4 (25 ng/ml), IFN-γ (100 ng/ml) and/or TNF-α (50 ng/ml) for 48 h, with 3–6 replicates of each treatment. Then, cell viability, post incubation with MTS reagent (one solution reagent) (20 μl/well) for 4 h, was assayed by monitoring the optical density (OD) at 490 nm using a Synergy™ 2 Multi-function Microplate Reader (Bio-Tek Instruments, Winooski, Vermont, USA). Live cells were recorded by counting viable cells using trypan blue exclusion.
2.5. Cell proliferation analysis and flow cytometry
Raji cells were seeded at density of 3×105 cells/well (for cell proliferation assay) and 2×106 cells/well (for flow cytometry) in 24-well and 6-well plates, respectively. Next day, cells were treated with hsBAFF (0–0.25 μg/ml) for 48 h. Subsequently, the number of proliferative cells was counted under a Coulter Counter (Beckman Coulter, Fullerton, CA, USA), and the ratios of live cells were monitored by a FACS Vantage SE flow cytometer (Beton Dickinson, California, USA) using Annexin-V-FITC/PI Apoptosis Detection kit.
2.6. Western blot analysis
Raji cells, or Raji cells infected with lentiviral shRNAs to S6K1, Erk1/2, S6K1/Erk1/2 and GFP, respectively were seeded in 6-well plate (2 × 106 cells/well) and cultured overnight in humidified incubator of 5% CO2 at 37°C. Next day, cells were treated with hsBAFF (0–0.25 μg/ml) for 12 h, or treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (5 and/or 50 ng/ml), IL-4 (5 and/or 25 ng/ml), IFN-γ (10 and/or 100 ng/ml) or TNF-α (5 and/or 50 ng/ml) for 12 h, or pre-incubated with/without U0126 (5 μM) for 1 h or rapamycin (0.2 μg/ml) for 2 h and then treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (50 ng/ml), IL-4 (25 ng/ml), IFN-γ (100 ng/ml) and/or TNF-α (50 ng/ml) for 12 h. Afterwards, total cell lysates were subjected to Western blotting as described previously [44]. The antibodies to phospho-Erk1/2 (Thr202/Tyr204), phosphor-S6K1 (Thr389) and phospho-S6 ribosomal protein (Ser235/236) were from Cell Signaling Technology (Beverly, MA, USA), whereas the antibodies to Erk2, S6K1, S6 ribosomal protein and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). G oat anti-rabbit IgG-horseradish peroxidase (HRP), goat anti-mouse IgG-HRP and rabbit anti-goat IgG-HRP were purchased from Pierce (Rockford, IL, USA).
2.7. Analysis of statistical significance
Results were presented as mean values ± standard error (mean ± S.E.). Analysis of statistical significance for indicated datasets was performed by using Student’s t-test for non-paired replicates. Group variability and interaction were compared using either one-way or two-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. A level of p < 0.05 was accepted to be significant.
3. Results
3.1. IL-2, IL-4, IFN-γ, or TNF-α reinforces proliferation and survival in hsBAFF-stimulated neoplastic B-lymphoid cells
To test whether or how cytokines IL-1, IL-4, IFN-γ and TNF-α affect proliferation and survival in hsBAFF-stimulated B cells, neoplastic B-lymphoid (Raji) cells were employed as a model to study the mechanisms. We have recently shown that administration of 0.5–5 μg/ml of hsBAFF for 48 h increased cell proliferation and survival in a dose-dependent fashion in Raji cells and primary B lymphocytes [40]. In agreement with the above findings, herein we also exhibited that treatment with 0.01–0.25 μg/ml hsBAFF for 48 h augmented cell proliferation and viability in Raji cells dose-dependently, as determined using cell counting (Fig. 1A) and MTS assay (Fig. 1B), respectively. Using trypan blue exclusive assay (Fig. 1C) and fluorescence-activated cell sorting (FACS) (Fig. 1D), we further showed that low-dose of hsBAFF also substantially elevated the relative number of live cells in the cells (Fig. 1C–D), indicating that the increase of cell survival by hsBAFF is related to the prolonged lifespan of the neoplastic B-lymphoid cells. Additionally, we noted that 0.25 μg/ml of hsBAFF was able to augment the cell proliferation/viability almost to a maximal level, thus this dose was chosen for more experiments, as described below.
Fig. 1.
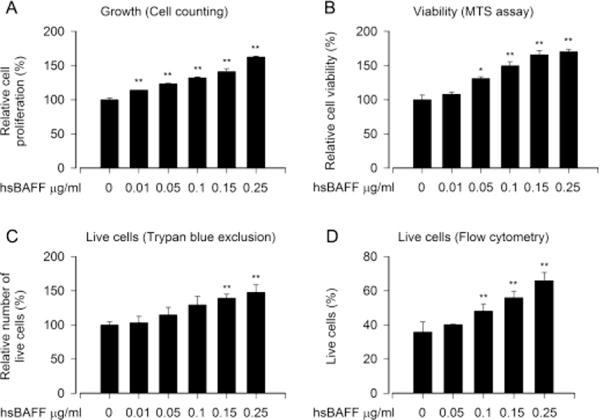
hsBAFF stimulates B-cell proliferation and survival in a concentration dependent manner. Raji cells were treated with 0–0.25 μg/ml hsBAFF for 48 h. (A) Cell proliferation was evaluated by cell counting. (B) Cell viability was monitored by MTS assay. (C) Relative number of live cells was estimated by trypan blue exclusion assay. (D) Quantitative live cells were calculated by FACS using annexin-V-FITC/PI staining. Results are presented as mean ± S.E. (n = 3–5). * p<0.05, ** p<0.01, difference with control group.
It is known that IL-2, IL-4, IFN-γ and/or TNF-α regulate immune response, including proliferation/viability, survival and Ig secretion, in normal and neoplastic B-lymphoid cells [22, 23, 25–30]. For this, Raji cells were treated with 0–100 ng/ml of IL-1, IL-4, IFN-γ or TNF-α alone for 48 h, respectively. As shown in Fig. 2A and B, a concentration-dependent increase of cell viability and survival induced by each cytokine was seen in the cells, and there existed a peak value for cell viability/survival at 50 ng/ml of IL-2 or TNF-α, 25 ng/ml of IL-4, and 100 ng/ml of IFN-γ. Next, we tested cell viability/survival induced by hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (5 and 50 ng/ml), IL-4 (5 and 25 ng/ml), IFN-γ (10 and 100 ng/ml) or TNF-α (5 and 50 ng/ml) in Raji cells. We found that IL-2, IL-4, IFN-γ or TNF-α potentiated the cell viability/survival stimulated by hsBAFF for 48 h in a concentration-dependent manner, compared to the control or hsBAFF group (Fig. 3A and B), respectively. The results suggest that IL-2/IL-4/IFN-γ/TNF-α can reinforce hsBAFF-stimulated viability/survival in neoplastic B-lymphoid cells.
Fig. 2.
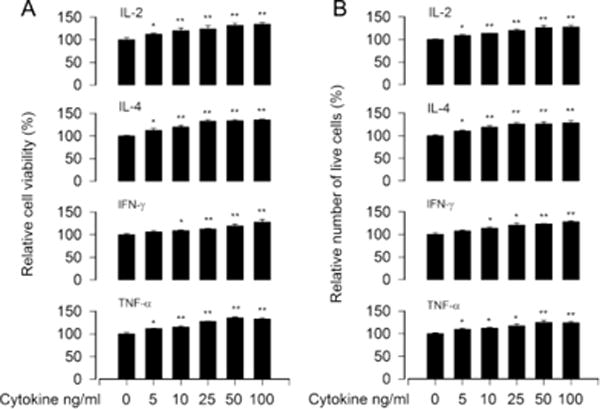
IL-2, IL-4, IFN-γ, or TNF-α promotes B-cell viability/survival. Raji cells were treated with 0–100 ng/ml of IL-2, IL-4, IFN-γ, or TNF-α for 48 h, respectively. (A) Cell viability was monitored by MTS assay. (B) Relative number of live cells was estimated by trypan blue exclusion assay. Results are presented as mean ± S.E. (n = 5). *p<0.05, **p<0.01, difference with control group.
Fig. 3.
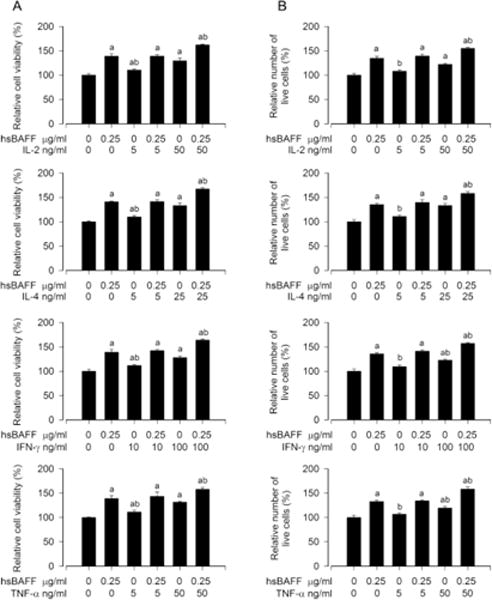
IL-2, IL-4, IFN-γ, or TNF-α strengthens hsBAFF-stimulated B-cell viability/survival. Raji cells were treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (5 and 50 ng/ml), IL-4 (5 and 25 ng/ml), IFN-γ (10 and 100 ng/ml), or TNF-α (5 and 50 ng/ml) for 48 h. (A) Cell viability was monitored by MTS assay. (B) Relative number of live cells was estimated by trypan blue exclusion assay. Results are presented as mean ± S.E. (n = 5). ap<0.05, difference with control group. bp<0.05, difference with 0.25 μg/ml hsBAFF group.
3.2. IL-2, IL-4, IFN-γ, or TNF-α strengthens hsBAFF-stimulated cell viability/survival through activating both Erk1/2 and S6K1 pathways in neoplastic B-lymphoid cells
Numerous studies have emphasized IL-2/IL-4/IFN-γ/TNF-α activation of several signaling pathways, including Erk1/2 and PI3K/Akt/S6K1 pathways [23, 38, 39]. Our group has recently shown that hsBAFF activates Erk1/2 contributing to cell proliferation/viability in normal and neoplastic B-lymphoid cells [40, 41]. hsBAFF elevates proliferation/survival in the cells, also in part, via activating mTOR-mediated S6K1 and 4E-BP1 pathways [41, 42]. This prompted us to search for the role of Erk1/2 and S6K1 activity in IL-2/IL-4/IFN-γ/TNF-α enhancement of hsBAFF-stimulated B-cell viability/survival. To gain insight into the molecular processes required for B-cell viability/survival, Raji cells were treated with different concentrations of hsBAFF (0–0.25 μg/ml) alone, or with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (5 and/or 50 ng/ml), IL-4 (5 and/or 25 ng/ml), IFN-γ (10 and/or 100 ng/ml), or TNF-α (5 and/or 50 ng/ml) for 12 h. Western blotting showed that hsBAFF obviously increased phospho-Erk1/2, phospho-S6K1 and phospho-S6 (a substrate of S6K1) dose-dependently (Fig. 4A and B). Treatment with IL-2, IL-4, IFN-γ or TNF-α alone elicited a dose-dependent elevation of phospho-Erk1/2, phospho-S6K1 and phospho-S6 as well (Fig. 4C and D). Of importance, addition of IL-2, IL-4, IFN-γ or TNF-α powerfully enhanced hsBAFF-induced phosphorylation of these proteins (Fig. 4C and D), suggesting that these cytokines can potentiate hsBAFF-activated Erk1/2 and S6K1 pathways in neoplastic B-lymphoid cells.
Fig. 4.
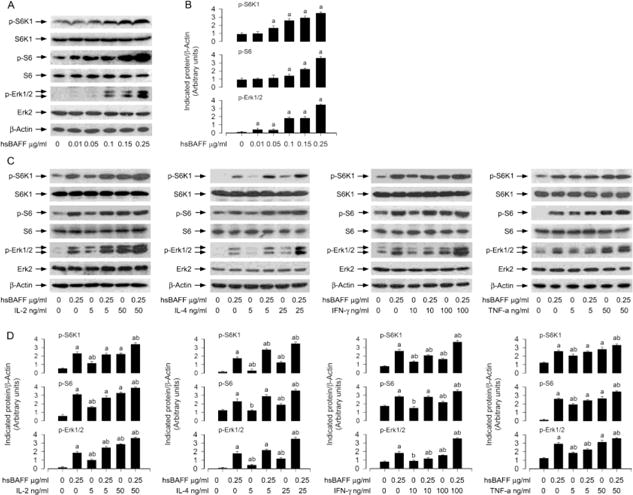
IL-2, IL-4, IFN-γ, or TNF-α reinforces hsBAFF-induced activation of Erk1/2 and S6K1 pathways in B cells. Raji cells were treated with hsBAFF (0–0.25 μg/ml) for 12 h, or with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (5 and 50 ng/ml), IL-4 (5 and 25 ng/ml), IFN-γ (10 and 100 ng/ml), or TNF-α (5 and 50 ng/ml) for 12 h. (A and C) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments (A and C), and the blots for p-Erk1/2, p-S6K1, and p-S6 were semi-quantified (B and D). Results are presented as mean ± S.E. (n = 3). ap<0.05, difference with control group. bp<0.05, difference with 0.25 μg/ml hsBAFF group.
3.3. Both Erk1/2 and S6K1 pathways are required for IL-2/IL-4/IFN-γ/TNF-α enhancement of viability/survival in hsBAFF-stimulated B cells
To further investigate the roles of Erk1/2 and S6K1 in IL-2/IL-4/IFN-γ/TNF-α enhancement of hsBAFF-stimulated B-cell viability/survival, pharmacological or genetic manipulation of the activity of Erk1/2 and S6K1 was conducted. At first, Raji cells were pretreated with/without U0126 (5 μM, a selective inhibitor of MKK1/2, upstream of Erk1/2) for 1 h and then treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (50 ng/ml), IL-4 (25 ng/ml), IFN-γ (100 ng/ml) or TNF-α (50 ng/ml) for 12 h or 48 h. We showed that U0126 almost completely suppressed the basal or hsBAFF-triggered phospho-Erk1/2 in the cells in response to each cytokine tested (Fig. 5A). Consistently, U0126 also significantly inhibited the basal or hsBAFF-induced B-cell viability/survival in the presence/absence of the individual cytokine, respectively, as detected by MTS (Fig. 5B) and trypan blue exclusion assay (Fig. 5C). To corroborate the above findings, genetic silence experiment for Erk1/2 was carried out. We showed that lentiviral shRNA to Erk1/2, but not to GFP, silenced expression of Erk1/2 protein by ~ 90% in Raji cells, as determined by Western blotting (Fig. 5D). Of importance, silencing Erk1/2 reduced the basal or the individual cytokine’s enhancement of viability/survival stimulated by hsBAFF (Fig. 5E and F), in the line with the effects of U0126 (Fig. 5B and C). These findings support the idea that Erk1/2 pathway is essential for IL-2/IL-4/IFN-γ/TNF-α potentiation of hsBAFF-stimulated B-cell viability/survival.
Fig. 5.
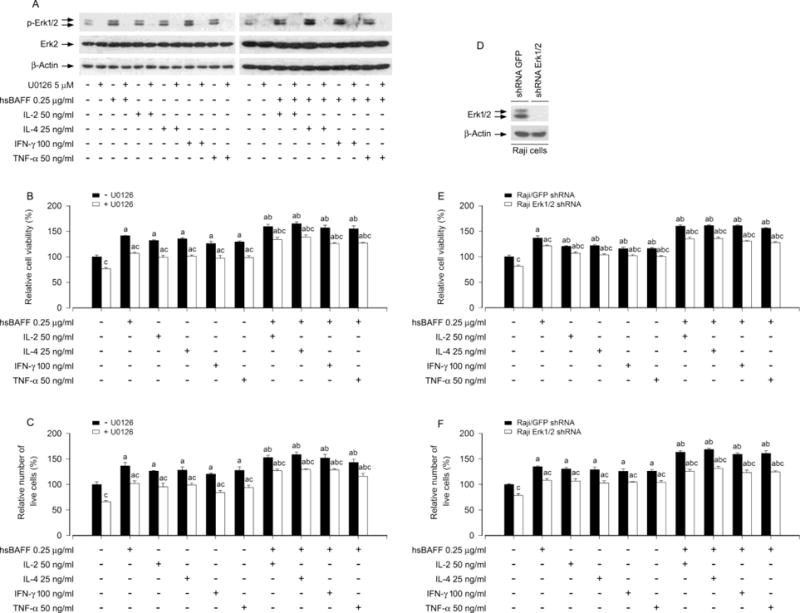
Pharmacological inhibition of Erk1/2 or down-regulation of Erk1/2 prevents hsBAFF-stimulated B-cell viability/survival enhanced by IL-2, IL-4, IFN-γ, or TNF-α. Raji cells, or Raji cells infected with lentiviral shRNAs to Erk1/2 and GFP, respectively, were pre-incubated with/without U0126 (5 μM) for 1 h and then treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (50 ng/ml), IL-4 (25 ng/ml), IFN-γ (100 ng/ml) or TNF-α (50 ng/ml) for 12 h (for Western blotting) or 48 h (for cell viability/survival assay). (A and D) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and E) Cell viability was monitored by MTS assay. (C and F) Relative number of live cells was estimated by trypan blue exclusion assay. Results are presented as mean ± S.E. (n = 5). ap < 0.05 difference with control group; bp < 0.05, difference with 0.25 μg/ml hsBAFF group; cp < 0.05 − U0126 group vs + U0126 group Erk1/2 shRNA group vs GFP shRNA group.
S6K1 is one of the best-characterized downstream effector molecules of mTORC1 [33, 34]. As rapamycin is a potent and specific inhibitor of mTORC1[35], we next employed rapamycin for inhibition of mTORC1-mediated S6K1 pathway. As expected, pretreatment with rapamycin resulted in complete blockage of the basal or hsBAFF-increased phospho-S6K1 and phospho-S6 in the cells treated with/without IL-2, IL-4, IFN-γ or TNF-α, as evidenced by Western blotting (Fig. 6A). In consistence with this, the basal or hsBAFF-enhanced B-cell viability/survival in the presence/absence of the individual cytokine was significantly attenuated by rapamycin as well (Fig. 6B and C). Next, we extended our studies using lentiviral shRNA to S6K1 to silence cellular protein expression of S6K1, showing that infection of Raji cells with lentiviral shRNA to S6K1 resulted in downregulation of cellular S6K1 protein by ~ 90%, compared to control cells infected with lentiviral shRNA to GFP (Fig. 6D). Of note, like treatment with rapamycin (Fig. 6B and C), silencing S6K1 significantly reduced the cell viability/survival stimulated by hsBAFF ± the individual cytokine in the cells (Fig. 6E and F). The data demonstrate that S6K1 pathway is as critical as Erk1/2 pathway for IL-2/IL-4/IFN-γ/TNF-α enhancement of hsBAFF-stimulated B-cell viability/survival.
Fig. 6.
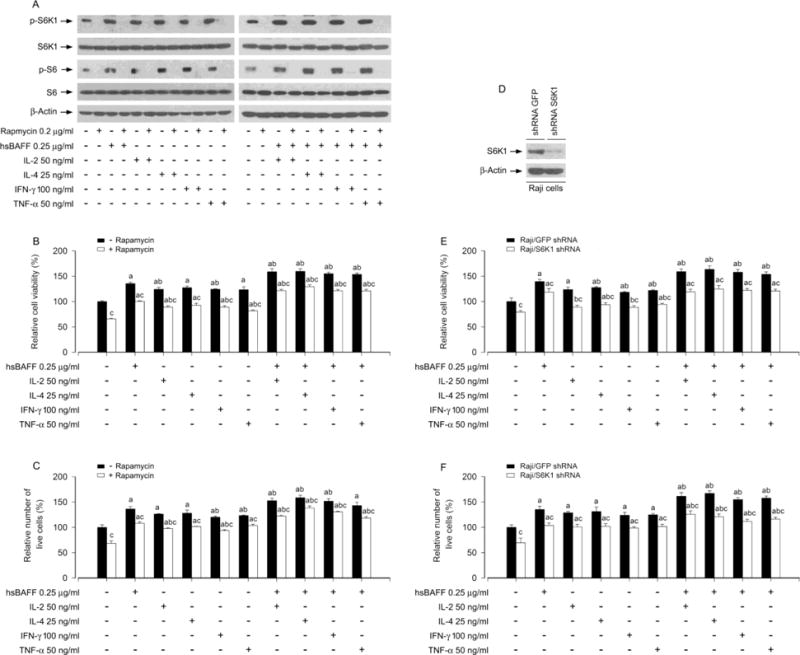
Pharmacological inhibition of S6K1 or down-regulation of S6K1 blocks hsBAFF-stimulated B-cell viability/survival enhanced by IL-2, IL-4, IFN-γ, or TNF-α. Raji cells, or Raji cells infected with lentiviral shRNAs to S6K1 and GFP, respectively, were pre-incubated with/without rapamycin (0.2 μg/ml) for 2 h and then treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (50 ng/ml), IL-4 (25 ng/ml), IFN-γ (100 ng/ml) or TNF-α (50 ng/ml) for 12 h (for Western blotting) or 48 h (for cell viability/survival assay). (A and D) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and E) Cell viability was monitored by MTS assay. (C and F) Relative number of live cells was estimated by trypan blue exclusion assay. Results are presented as mean ± S.E. (n = 5). ap < 0.05 difference with control group; bp < 0.05, difference with 0.25 μg/ml hsBAFF group; cp < 0.05 − Rapamycin group vs + Rapamycin group or S6K1 shRNA group vs GFP shRNA group.
Next, we queried whether IL-2/IL-4/IFN-γ/TNF-α enhancement of viability/survival in hsBAFF-stimulated B cells is indeed by activating both Erk1/2 and S6K1 pathways. To answer this question, we worked on our studies by double knockdown of Erk1/2 and S6K1. Western blot results revealed that co-infection with lentiviral shRNAs to Erk1/2 and S6K1silenced S6K1/Erk1/2 protein expression by ~90% in Raji cells (Fig. 7A). Of interest, the double silencing of S6K1/Erk1/2 showed a more potent inhibitory effect on the cell viability/survival stimulated by hsBAFF ± the individual cytokine in the cells (Fig. 7B and C), compared to the single silencing of Erk1/2 or S6K1 in the cells (Fig. 5 and 6). Collectively, the results underline that both Erk1/2 and S6K1 pathways are required for IL-2/IL-4/IFN-γ/TNF-α enhancement of viability/survival in hsBAFF-stimulated B cells.
Fig. 7.
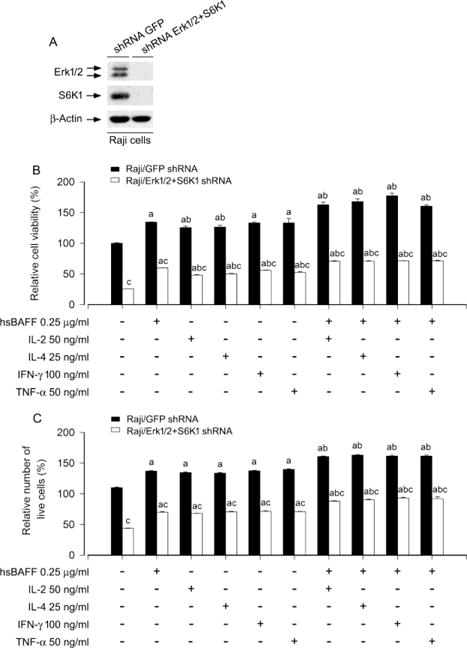
Double silencing of Erk1/2 and S6K1 powerfully inhibits hsBAFF-stimulated B-cell viability/survival enhanced by IL-2, IL-4, IFN-γ, or TNF-α. Raji cells were infected with lentiviral shRNAs to S6K1/Erk1/2 and GFP, respectively. (A) The cells were subjected to Western blotting. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and C) The cells were treated with/without hsBAFF (0.25 μg/ml) in the presence or absence of IL-2 (50 ng/ml), IL-4 (25 ng/ml), IFN-γ (100 ng/ml) or TNF-α (50 ng/ml) for 48 h, followed by cell viability using MTS assay (B) and relative number of live cells using trypan blue exclusion assay (C). Results are presented as mean ± S.E. (n = 5). ap < 0.05 difference with control group; bp < 0.05, difference with 0.25 μg/ml hsBAFF group; cp < 0.05 S6K1/Erk1/2 shRNA group vs GFP shRNA group.
4. Discussion
A series of data have uncovered that BAFF acts as a crucial factor in aggressive B-cell malignancies and various autoimmune diseases, such as SLE, RA and SS [13, 21, 41, 46, 47]. For example, lupus serum contains elevated levels of BAFF [48]. High levels of BAFF evoke prolonged lifespan B-cell disorders [49, 50]. The SLE patients suffer from numerous pathogenic autoantibodies released from aggressive B cells, due to excessive BAFF [17, 21, 46]. However, abnormal release or effects of diverse other cytokines have been also documented in SLE patients and animal models both in vitro and in vivo, pointing to association of the autoimmune disease with the dysregulation of complex cytokine networks [47, 51]. It has been reported that IL-2, IL-4, IFN-γ and/or TNF-α regulate immune response, including proliferation/viability, survival and Ig secretion, in normal and neoplastic B-lymphoid cells [22, 23, 25–30]. IL-2 dysregulation is considered as a possible factor for lupus pathogenesis [51]. Serum IFN-γ level is elevated in some SLE patients [51, 52]. IL-2, IL-4, and IFN-γ act as pro-survival factors in B-cell chronic lymphocytic leukaemia (B-CLL) and prevent the leukemic cells from undergoing apoptosis [30]. There exists a potential regulative role of TNF-α on aggressive B-cell disorders [26–30]. The data suggest that uncontrolled B-cell activation and autoimmune diseases (e.g. SLE) are substantially provoked by cytokine IL-2, IL-4, IFN-γ or TNF-α. Thus, it is of great importance to understand whether or how IL-2, IL-4, IFN-γ or TNF-α affects BAFF-stimulated neoplastic B-lymphoid cells relative to autoimmune diseases. In this study, we found that treatment with IL-2, IL-4, IFN-γ, or TNF-α increased hsBAFF-induced cell proliferation and survival concentration-dependently in Raji cells. The results suggest that IL-2, IL-4, IFN- γ, or TNF-α may potently enhance BAFF-induced aggressive B-cell malignancies.
Studies have shown that IL-2, IL-4, IFN-γ, or TNF-α activates Erk1/2 and PI3K/Akt/S6K1 pathways [23, 38, 39]. Our group has recently revealed that hsBAFF enhances cell proliferation/viability in Erk1/2-dependent manner in normal and neoplastic B-lymphoid cells [40, 41]. Moreover, hsBAFF also augments proliferation/survival in the cells, at least in part, through activating mTOR-mediated S6K1 and 4E-BP1 pathways [41, 42]. Here, we identified that IL-2, IL-4, IFN-γ or TNF-α reinforced hsBAFF-stimulated B-cell viability/survival by activating both Erk1/2 and S6K1 pathways. This is supported by the findings that U0126 or rapamycin inhibited the phosphorylation of Erk1/2 or S6K1 contributing to significant reduction of viability/survival in Raji cells treated with hsBAFF ± IL-2, IL-4, IFN-γor TNF-α, respectively (Fig. 5 and 6). Further, we observed that single or double knockdown of Erk1/2 and S6K1 by using lentivial shRNAs to Erk1/2 and S6K1 attenuated the cell viability/survival stimulated by hsBAFF ± the individual cytokine in Raji cells (Fig. 5–7), and especially there existed a more potent decline for the double S6K1/Erk1/2-silenced cells than for the single S6K1 or Erk1/2-silenced cells. To our knowledge, this is the first report presenting that the cytokines IL-2/IL-4/IFN-γ/TNF-α enhance BAFF-stimulated proliferation and survival by activating Erk1/2 and S6K1 signaling pathways in neoplastic B-lymphoid cells.
As BAFF extends B-cell lifespan in excess of physiological limits [17–21], the malignant B cells with prolonged lifespan have been demonstrated to exert central roles in the pathogenesis of association B-cell disorders with autoimmune diseases [21]. Notably, multiple studies have documented that T cell-dependent B-cell secretion of autoantibody lies at the heart of pathogenesis for autoimmune disease (e.g. SLE) [52]. Cytokines that activate B and T cells and promote their interaction forms key drivers related to the autoimmune disease [52]. These findings imply that manipulation of key cytokines can likely provide important clues to the pathogenic mechanisms underlying specific forms of autoimmune diseases, and pave the way toward more effective therapeutics. In this study, we observed the enhanced effects of IL-2, IL-4, IFN-γ or TNF-α on proliferation and survival in hsBAFF-stimulated Raji B-lymphoid cells (Fig. 3). We also demonstrated that both Erk1/2 and S6K1 pathways were required for IL-2, IL-4, IFN-γ or TNF-α’s enhancement of viability/survival in the cells triggered by hsBAFF (Fig. 5–7). A proposition that arises from this work is that IL-2, IL-4, IFN-γ or TNF-α’s enhancement of BAFF-activated Erk1/2 and S6K1 signaling may be an important mechanism in aggressive or neoplastic B-cells and autoimmune B cells. Our results expand a conceptual view of BAFF signaling associated with complex cytokine networks, which is involved in the pathophysiology of aggressive or neoplastic B-cell disorders.
In conclusion, we have identified that hsBAFF-promoted cell viability/survival was enhanced by treatment with IL-2, IL-4, IFN-γ or TNF-α in Raji cells. Mechanistically, the individual cytokine enhances BAFF-stimulated viability/survival by activating Erk1/2 and S6K1 pathways in the cells. Our findings underline that manipulation of IL-2, IL-4, IFN-γ and/or TNF-α levels, or inhibitors of Erk1/2 or S6K1 may be a new approach to prevent BAFF-induced aggressive B-cell malignancies related to autoimmune diseases.
Highlights.
IL-2/IL-4/IFN-γ/TNF-α reinforces BAFF-stimulated viability/survival in neoplastic B-lymphoid cells.
IL-2/IL-4/IFN-γ/TNF-α enhances BAFF-stimulated B-cell viability/survival by activating Erk1/2 and S6K1 pathways.
Manipulating IL-2/IL-4/IFN-γ/TNF-α level or blocking Erk1/2 or S6K1 may prevent BAFF-induced B-cell malignancies.
Acknowledgments
This work was supported in part by the grants from National Natural Science Foundation of China (No.31172083; L.C.), NIH (CA115414; S.H.), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD-14KJB180010; L.C.), American Cancer Society (RSG-08-135-01-CNE; S.H.), Louisiana Board of Regents (NSF-2009-PFUND-144; S.H.), and Innovative Research Program of Jiangsu College Graduate of China (No. CXLX13_387; L.G.).
Abbreviations
- 4E-BP1
Eukaryotic initiation factor 4E binding protein 1
- Akt
protein kinase B (PKB)
- BAFF
B-cell activating factor of the TNF family
- BLyS
B lymphocyte stimulator
- BCMA
B cell maturation antigen
- Erk1/2
extracellular signal-related kinase 1/2
- IFN
interferon
- IL
interleukin
- MAPK
mitogen-activated protein kinase
- MKK
mitogen-activated protein kinase kinase
- mTOR
mammalian target of rapamycin
- PI3K
phosphatidylinositol 3′-kinase
- RA
rheumatoid arthritis
- S6K1
S6 kinase 1
- SLE
systemic lupus erythematosus
- SS
Sjögren’s syndrome
- TACI
transmembrane activator and cyclophilin ligand interactor
- TALL-1
TNF and apoptosis ligand-related leukocyte-expressed ligand 1
- THANK
TNF homologue that activates apoptosis, nuclear factor κB, and c-Jun NH2-terminal kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Henley T, Kovesdi D, Turner M. B-cell responses to B-cell activation factor of the TNF family (BAFF) are impaired in the absence of PI3K delta. Eur J Immunol. 2008;38:3543–8. doi: 10.1002/eji.200838618. [DOI] [PubMed] [Google Scholar]
- 2.Mueller CG, Boix C, Kwan WH, Daussy C, Fournier E, Fridman WH, et al. Critical role of monocytes to support normal B cell and diffuse large B cell lymphoma survival and proliferation. J Leukoc Biol. 2007;82:567–75. doi: 10.1189/jlb.0706481. [DOI] [PubMed] [Google Scholar]
- 3.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–56. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–3. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 5.Shu HB, Hu WH, Johnson H. TALL-1 is a novel member of the TNF family that is down-regulated by mitogens. J Leukoc Biol. 1999;65:680–3. [PubMed] [Google Scholar]
- 6.Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol. 2006;18:263–75. doi: 10.1016/j.smim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–64. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 8.Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–9. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- 10.Lavie F, Miceli-Richard C, Quillard J, Roux S, Leclerc P, Mariette X. Expression of BAFF (BLyS) in T cells infiltrating labial salivary glands from patients with Sjogren’s syndrome. J Pathol. 2004;202:496–502. doi: 10.1002/path.1533. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Park CS, Yoon SO, Li L, Hsu YM, Ambrose C, et al. BAFF supports human B cell differentiation in the lymphoid follicles through distinct receptors. Int Immunol. 2005;17:779–88. doi: 10.1093/intimm/dxh259. [DOI] [PubMed] [Google Scholar]
- 12.Schneider P, Tschopp J. BAFF and the regulation of B cell survival. Immunol Lett. 2003;88:57–62. doi: 10.1016/s0165-2478(03)00050-6. [DOI] [PubMed] [Google Scholar]
- 13.Mackay F, Silveira PA, Brink R. B cells and the BAFF/APRIL axis: fast-forward on autoimmunity and signaling. Curr Opin Immunol. 2007;19:327–36. doi: 10.1016/j.coi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Fu L, Lin-Lee YC, Pham LV, Tamayo AT, Yoshimura LC, Ford RJ. BAFF-R promotes cell proliferation and survival through interaction with IKKbeta and NF-kappaB/c-Rel in the nucleus of normal and neoplastic B-lymphoid cells. Blood. 2009;113:4627–36. doi: 10.1182/blood-2008-10-183467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baker KP. BLyS–an essential survival factor for B cells: basic biology, links to pathology and therapeutic target. Autoimmun Rev. 2004;3:368–75. doi: 10.1016/j.autrev.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Stewart DM, McAvoy MJ, Hilbert DM, Nelson DL. B lymphocytes from individuals with common variable immunodeficiency respond to B lymphocyte stimulator (BLyS protein) in vitro. Clin Immunol. 2003;109:137–43. doi: 10.1016/s1521-6616(03)00215-8. [DOI] [PubMed] [Google Scholar]
- 17.Moisini I, Davidson A. BAFF: a local and systemic target in autoimmune diseases. Clin Exp Immunol. 2009;158:155–63. doi: 10.1111/j.1365-2249.2009.04007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bosello S, Youinou P, Daridon C, Tolusso B, Bendaoud B, Pietrapertosa D, et al. Concentrations of BAFF correlate with autoantibody levels, clinical disease activity, and response to treatment in early rheumatoid arthritis. J Rheumatol. 2008;35:1256–64. [PubMed] [Google Scholar]
- 19.Zhang J, Roschke V, Baker KP, Wang Z, Alarcon GS, Fessler BJ, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166:6–10. doi: 10.4049/jimmunol.166.1.6. [DOI] [PubMed] [Google Scholar]
- 20.Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–85. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- 21.Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–37. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mingari MC, Gerosa F, Carra G, Accolla RS, Moretta A, Zubler RH, et al. Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature. 1984;312:641–3. doi: 10.1038/312641a0. [DOI] [PubMed] [Google Scholar]
- 23.Le Gallou S, Caron G, Delaloy C, Rossille D, Tarte K, Fest T. IL-2 requirement for human plasma cell generation: coupling differentiation and proliferation by enhancing MAPK-ERK signaling. J Immunol. 2012;189:161–73. doi: 10.4049/jimmunol.1200301. [DOI] [PubMed] [Google Scholar]
- 24.Mehta DS, Wurster AL, Whitters MJ, Young DA, Collins M, Grusby MJ. IL-21 induces the apoptosis of resting and activated primary B cells. J Immunol. 2003;170:4111–8. doi: 10.4049/jimmunol.170.8.4111. [DOI] [PubMed] [Google Scholar]
- 25.Snapper CM, Paul WE. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. 1987;236:944–7. doi: 10.1126/science.3107127. [DOI] [PubMed] [Google Scholar]
- 26.Kehrl JH, Miller A, Fauci AS. Effect of tumor necrosis factor alpha on mitogen-activated human B cells. J Exp Med. 1987;166:786–91. doi: 10.1084/jem.166.3.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cordingley FT, Bianchi A, Hoffbrand AV, Reittie JE, Heslop HE, Vyakarnam A, et al. Tumour necrosis factor as an autocrine tumour growth factor for chronic B-cell malignancies. Lancet. 1988;1:969–71. doi: 10.1016/s0140-6736(88)91782-5. [DOI] [PubMed] [Google Scholar]
- 28.Digel W, Stefanic M, Schoniger W, Buck C, Raghavachar A, Frickhofen N, et al. Tumor necrosis factor induces proliferation of neoplastic B cells from chronic lymphocytic leukemia. Blood. 1989;73:1242–6. [PubMed] [Google Scholar]
- 29.Foa R, Massaia M, Cardona S, Tos AG, Bianchi A, Attisano C, et al. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukemia cells: a possible regulatory role of TNF in the progression of the disease. Blood. 1990;76:393–400. [PubMed] [Google Scholar]
- 30.Mainou-Fowler T, Miller S, Proctor SJ, Dickinson AM. The levels of TNF alpha, IL4 and IL10 production by T-cells in B-cell chronic lymphocytic leukaemia (B-CLL) Leuk Res. 2001;25:157–63. doi: 10.1016/s0145-2126(00)00097-7. [DOI] [PubMed] [Google Scholar]
- 31.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 32.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–43. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013;23:53–62. doi: 10.1016/j.gde.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anti-cancer agents in medicinal chemistry. 2010;10:571–581. doi: 10.2174/187152010793498663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang S, Houghton PJ. Targeting mTOR signaling for cancer therapy. Current opinion in pharmacology. 2003;3:371–377. doi: 10.1016/s1471-4892(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 37.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15:155–62. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- 38.Matto M, Nuutinen UM, Ropponen A, Myllykangas K, Pelkonen J. CD45RA and RO isoforms have distinct effects on cytokine- and B-cell-receptor-mediated signalling in human B cells. Scand J Immunol. 2005;61:520–8. doi: 10.1111/j.1365-3083.2005.01624.x. [DOI] [PubMed] [Google Scholar]
- 39.Kim HP, Imbert J, Leonard WJ. Both integrated and differential regulation of components of the IL-2/IL-2 receptor system. Cytokine Growth Factor Rev. 2006;17:349–66. doi: 10.1016/j.cytogfr.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Liang D, Zeng Q, Xu Z, Zhang H, Gui L, Xu C, et al. BAFF activates Erk1/2 promoting cell proliferation and survival by Ca2+-CaMKII-dependent inhibition of PP2A in normal and neoplastic B-lymphoid cells. Biochem Pharmacol. 2014;87:332–43. doi: 10.1016/j.bcp.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng Q, Zhang H, Qin J, Xu Z, Gui L, Liu B, et al. Rapamycin inhibits BAFF-stimulated cell proliferation and survival by suppressing mTOR-mediated PP2A-Erk1/2 signaling pathway in normal and neoplastic B-lymphoid cells. Cell Mol Life Sci. 2015;72:4867–84. doi: 10.1007/s00018-015-1976-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ke Z, Liang D, Zeng Q, Ren Q, Ma H, Gui L, et al. hsBAFF promotes proliferation and survival in cultured B lymphocytes via calcium signaling activation of mTOR pathway. Cytokine. 2013;62:310–21. doi: 10.1016/j.cyto.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 43.Cao P, Mei JJ, Diao ZY, Zhang S. Expression, refolding, and characterization of human soluble BAFF synthesized in Escherichia coli. Protein Expr Purif. 2005;41:199–206. doi: 10.1016/j.pep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Liu L, Luo Y, Huang S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J Neurochem. 2008;105:251–61. doi: 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- 45.Liu L, Li F, Cardelli J, Martin K, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- 46.Kaneko T, Amano H, Kawano S, Minowa K, Ando S, Watanabe T, et al. Increased serum concentration of BAFF/APRIL and IgA2 subclass in patients with mixed connective tissue disease complicated by interstitial lung disease. Mod Rheumatol. 2014;24:310–5. doi: 10.3109/14397595.2013.843748. [DOI] [PubMed] [Google Scholar]
- 47.Su DL, Lu ZM, Shen MN, Li X, Sun LY. Roles of pro- and anti-inflammatory cytokines in the pathogenesis of SLE. J Biomed Biotechnol. 2012;2012:347141. doi: 10.1155/2012/347141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davidson A. Targeting BAFF in autoimmunity. Curr Opin Immunol. 2010;22:732–9. doi: 10.1016/j.coi.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cornec D, Devauchelle-Pensec V, Tobon GJ, Pers JO, Jousse-Joulin S, Saraux A. B cells in Sjogren’s syndrome: from pathophysiology to diagnosis and treatment. J Autoimmun. 2012;39:161–7. doi: 10.1016/j.jaut.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 50.Ramanujam M, Davidson A. BAFF blockade for systemic lupus erythematosus: will the promise be fulfilled? Immunol Rev. 2008;223:156–74. doi: 10.1111/j.1600-065X.2008.00625.x. [DOI] [PubMed] [Google Scholar]
- 51.Apostolidis SA, Lieberman LA, Kis-Toth K, Crispin JC, Tsokos GC. The dysregulation of cytokine networks in systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31:769–79. doi: 10.1089/jir.2011.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis LS, Hutcheson J, Mohan C. The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31:781–9. doi: 10.1089/jir.2011.0047. [DOI] [PMC free article] [PubMed] [Google Scholar]