

Abstract
The human neuropeptide Y4 receptor (Y4R) and its native ligand, pancreatic polypeptide, are critically involved in the regulation of human metabolism by signaling satiety and regulating food intake, as well as increasing energy expenditure. Thus, this receptor represents a putative target for treatment of obesity. With respect to new approaches to treat complex metabolic disorders, especially in multi-receptor systems, small molecule allosteric modulators have been in the focus of research in the last years. However, no positive allosteric modulators or agonists of the Y4R have been described so far. In this study, small molecule compounds derived from the Niclosamide scaffold were identified by high-throughput screening to increase Y4R activity. Compounds were characterized for their potency and their effects at the human Y4R and as well as their selectivity towards Y1R, Y2R and Y5R. These compounds provide a structure-activity relationship profile around this common scaffold and lay the groundwork for hit-to-lead optimization and characterization of positive allosteric modulators of the Y4R.
Introduction
Obesity, a major risk factor for diabetes, heart disease, cancer, and mortality is a rising medical concern with doubled worldwide prevalence since 1980, reaching an estimated medical cost of $147 billion in 2008 [1, 2]. Dietary changes and nutritional counseling can be effective treatment options but results are inconsistent, suffering from poor long-term patient adherence often leading to weight regain [3]. So far, only invasive treatments such as bariatric surgery show long term success rates, but are limited to patients where the benefits outweigh the risks and costs [4]. Several studies suggest that hormonal changes following bariatric surgery contribute to its long term success [2, 5]. Their respective hormone receptors may represent promising therapeutic targets. For example, meal-stimulated glucagon-like peptide-1 (GLP-1) release is thought to participate in the long-term success of bariatric procedures. GLP-1 receptor agonists have been shown to produce weight loss and glucose homeostasis for subjects with type II diabetes. However, the weight loss seen with GLP-1 agonists alone is modest [6].
Two members of the pancreatic polypeptide family including peptide tyrosine tyrosine (PYY) and pancreatic polypeptide (PP) act as satiety factors to inhibit food intake, and modify metabolic homeostasis [7]. Along with the third member of this class of hormones, neuropeptide Y (NPY), the peptides regulate energy metabolism through four different Y receptor subtypes in humans: Y1R, Y2R, Y4R and Y5R. All receptor subtypes are involved in the regulation of energy metabolism and are putative targets for the treatment of obesity. Furthermore, in this multi ligand/multi receptor system, the receptor subtypes display different preferences for NPY, PYY and PP. Y1R, Y2R and Y5R bind NPY and PYY with high affinity. In contrast, PP is strongly preferred by the Y4R and binds to this receptor subtype with a high affinity and with lower affinity to the Y5R [8].
While PP and PYY both present promising routes for the treatment of obesity, PP may be preferred as it inhibits feeding in mice more than PYY and PYY-3-36 [9]. Pancreatic polypeptide has also been shown to inhibit food intake in humans [10]. Further, in contrast to PP, medically relevant doses of PYY induce nausea in humans [10, 11].
PP is released under vagal cholinergic control from F-cells of pancreatic islets in response and proportion to food ingestion [12]. The hormone is furthermore expressed in some endocrine cells of the intestines [13]. Primarily through Y4R, PP promotes appetite suppression, inhibition of gastric emptying, and increased energy expenditure [14]. The human Y4R is a 375 amino acid class A G-protein coupled receptor (GPCR) primarily expressed in the gastrointestinal tract, where it inhibits peristalsis and excretion [15]. Other peripheral organs that express Y4R include the heart, skeletal muscle, and thyroid gland. In the central nervous system, Y4R is expressed in the hypothalamus, where it relays anorexigenic signals [16] and inhibits neurotransmitter release [17]. The Y4R is a putative target for the treatment of obesity based on its strong anorexigenic potential and studies involving the overexpression or endogenous application of PP [16, 18–20].
Our efforts focus on the identification of small-molecule positive allosteric modulators (PAMs) of Y4R. Allosteric ligands represent promising options for treatment of metabolic and neurological diseases [21]. Allosteric ligands show a range of pharmacological activities including PAMs (agonism, potentiation or both), negative allosteric modulation (NAM), and inverse agonism. These ligands have the potential to differentially regulate several pathways on the same GPCR and induce a biased signaling [22]. Additionally, PAMs with little or no intrinsic activity may be safer therapeutics because their dependence on the presence of the endogenous agonist may help to prevent toxicity and other negative side effects [23]. This approach preserves the physiological signaling patterns, which may be critical in complex systems and is not feasible using orthosteric agonists [24]. Allosteric binding sites may be less conserved between receptor subtypes than the orthosteric binding site because they lack the evolutionary pressure to conserve affinity for the orthosteric ligand. Therefore it is often possible to design allosteric modulators with high selectivity [24, 25].
Since no small-molecule PAM or agonist has been identified for Y4R, we used high-throughput screening (HTS) [26] to identify compounds that modulate the Y4R. Initial hit compounds were validated as PAMs in a complementary set of assays and subtype-selectivity was investigated for all human NPY receptors.
Materials and Methods
Cell Culture
COS-7 cells stably expressing hY1/2/4/5R_eYFP fusion protein and the Δ6Gαqi4myr chimeric Gα-protein were prepared as previously described [27]. The hY1/2/4/5R_eYFP cDNA was subcloned into MCS1 of a pVitro2-MCS vector carrying a hygromycin resistance gene; the Δ6Gαqi4myr cDNA was subcloned into MCS1 of a pVitro2-MCS vector carrying a G418 resistance gene (Invivogen, San Diego, CA).
COS-7 (African Green Monkey kidney) cells stably expressing the hY1/2/4/5R_eYFP fusion protein and the Δ6Gαqi4myr chimeric Gα-protein were cultured at 37°C in high glucose Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies, Carlsbad, CA) with glutamine and sodium pyruvate (Life Technologies/Lonza) supplemented with 10% (w/v) FBS (Invitrogen, Carlsbad, CA), 1.5 mg/mL G418-sulfate (Amresco, Solon, OH) and 133 μg/ml hygromycin (Invivogen).
HTS Calcium Flux Assay
The purities of compounds in the Vanderbilt HTS facility are more than 95% as confirmed by the supplier. A total of 35,288 compounds were tested for their ability to modulate the Y4R activation in conjunction with the Vanderbilt HTS facility. In a pilot HTS experiment, 2000 compounds (spectrum collection, MicroSource Discovery Systems, Inc., Gaylordsville, CT) were screened for modulation of the Y4R. This collection is designed to enrich the general hit rate by including drugs with known biological profiles (60%), naturally occurring products with no biological profile (25%), and non-drug compounds with biological profiles (15%). In a following HTS, 33,288 compounds were tested for Y4R modulatory effects. Thirty-two thousand of these compounds were randomly selected from the Vanderbilt compound library and 1288 were selected based on their similarity to Niclosamide, a PAM discovered in the pilot HTS.
Cells were plated in TC-treated 384-well plates (black, clear bottom, Greiner, Monroe, NC) in 20 μL cell culture medium using a Multidrop Combi (Thermo Fisher, Waltham, MA) microplate dispenser (ThermoScientific, Thermo Fisher) at a density of 16,000 cells/well. The cells were incubated for 24 hours at 37°C in the presence of 5% CO2. Following incubation, the medium was replaced with 20 μL/well fluorescent dye solution (1.0 μM Fluo-2 AM (TEFlabs, Austin, TX), .01% (v/v) Pluronic Acid F-127 in assay buffer) using an ELx405 cell washer (BioTek, Winooski, VT). Following a 90 minute incubation at room temperature, fluorescent dye solution was replaced with 20 μL/well assay buffer (HBSS, 20 mM HEPES, and 1.25 mM Probenecid (Sigma-Aldrich, St. Louis, MO)) using the ELx405 and cell plates were loaded into a Functional Drug Screening System (FDSS, Hamamatsu, Japan). Once loaded into the FDSS, cell plates were imaged at 1Hz (excitation 470 ± 20 nm, emission 540 ± 30 nm using a 3-addition protocol designed to detect agonists, potentiators, and inhibitors: 1) after collecting 4 seconds of baseline, 20 μl/well of 20 μM test compounds in assay buffer + 0.1% (w/v) fatty-acid-free bovine serum albumin (Sigma-Aldrich, modified assay buffer) were added 2) following a 150 second delay, 10 μl/well of concentration of 5-fold over the PP EC20 (55 ± 27 pM) 3) after 330 seconds, a 13 μl/well addition 5-fold over the PP EC80 (836 ± 33 pM) in modified assay buffer was performed. On each screening day, PP EC20 and EC80 plates were adjusted after a test PP CRC at the beginning of each day to account for minor day to day variations in experimental conditions.
Substructure Search
Substructure searches were performed against the Vanderbilt Institute for Chemical Biology (VICB) library using the ChemCart application (DeltaSoft Inc., Hillsborough, NJ) Tanimoto coefficient similarity search. Higher Tanimoto coefficients indicate more similarity based on the shared presence of chemical subgroups. To increase our chemical search space, we altered the amide linker of Niclosamide and repeated the substructure search against the VICB library. Linker alterations included replacing the amide linker with urea, thiourea, δ-lactam, and an extension of the linker by one or two methylene groups. Tanimoto coefficient cut-offs were adjusted to between 0.45 and 0.63 for each substructure search to ensure that approximately 200 to 300 compounds were identified for each scaffold (S1 Table). Reference structures with alternate backbone constitutions were generated using ChemBioDraw Ultra (PerkinElmer Inc., Waltham, MA). Final search results were concatenated and duplicate search hits were removed. The final collection of 1288 compounds were distributed over five plates and tested using the triple-add screen protocol.
IP3 Assay
Cells were seeded into 48-well plates and incubated for 24 hours at 37°C / 5% CO2. Afterwards, cells were labeled for at least 16 hours in DMEM+10% (w/v) FBS containing 2 μCi/ml myo-[2- 3H(N)]-inositol (PerkinElmer) at 37°C / 5% CO2. Labeling solution was aspirated and cells were washed with 20 μL/well DMEM + 10 mM LiCl (Sigma-Aldrich; DMEM/LiCl) and stimulated with the peptide solutions and compounds. Specifically, 50 μl/well DMEM/LiCl were added after washing, followed by addition of 50 μl/well test compound in DMEM/LiCl (3-fold over the final concentration) and addition of 50 μl/well peptide solution (3-fold over the final concentration) in DMEM/LiCl. Stimulation was performed for 2 hours at 37°C / 5% CO2. Cell lysis, subsequent sample preparation and radiometric detection was performed as described previously [27].
Data analysis
Data analysis was performed with GraphPad Prism 5.03 software (GraphPad Software, San Diego, CA). Calculation of EC50 and pEC50 was performed using standard non-linear regression (log(agonist) vs. response, three parameters). All data were normalized to the corresponding control curve in the absence of the modulator. Final concentration response curves were created by calculating the mean and SEM of all individual experiments for each data point. Statistical evaluation was performed using two-way ANOVA and Bonferroni post test with * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Results
Identification of Y4R PAMs
Until now, no small molecule agonists of Y receptors have been described. Based on this lack of any structure activity data, identification of Y4R PAMs was initiated with a Ca2+-flux-based HTS approach. An initial pilot screen was performed with the ‘Spectrum collection’, a small scale library with 2000 compounds comprising synthetic small molecules as well as purified natural products covering a range of known biologically active properties. This pilot screen yielded 65 putative PAMs. All initial hits were retested for off-target effects in wildtype COS-7 cells and the PAM effect was validated via concentration-dependent potentiation of a submaximal PP response. After eliminating structures that show off-target effects, seven compounds with potencies in the micromolar range were identified (S1 Fig) for further investigation. Validation of the Y4R PAM activity was performed with a well-established assay system for Y receptor activation studies, based on cellular accumulation of inositol phosphate [28, 29]. Furthermore, the compound activity at Y1R, Y2R and Y5R was examined at this stage of Y4R PAM identification. Therefore, Y receptor activation with an agonist ligand concentration causing a submaximal response (1 nM, Fig 1) was monitored in presence and absence of test compound. This experimental setup allows the detection of potential compound-induced shifts of the concentration-response curve and parallel testing of all compounds on all Y receptor subtypes. Cells stably expressing the chimeric G-protein Δ6Gαqi4myr and one of four different human NPY receptor subtypes (Y1R, Y2R, Y4R, or Y5R) were treated with test compound at a final concentration of 10 μM followed immediately by stimulation with 1 nM endogenous peptide agonist (Y4R: PP, Y1,2,5R: NPY).
Fig 1. Validation of Y4R PAM activity and subtype selectivity of initial Ca2+-flux-based screen hit compounds in an inositol phosphate accumulation assay.

(A) Compound structures. (B) Effect of 10 μM compound on submaximal YR activation by 1 nM ligand, which represents EC20-EC60 (Y1,2,5R: NPY; Y4R: PP). Data represent the mean ± SEM of two independent experiments each performed in quadruplicate (*** p ≤ .001 Bonferroni).
The effect of the compounds on inositol phosphate accumulation was investigated in relation to the control in presence of DMSO (DMSO set to 100%, complete data of all 7 compounds see S1 Fig). However, only three of the seven HTS PAM hits validated their Y4R PAM effect also in the inositol phosphate accumulation assay (Fig 1). Of all tested compounds, Niclosamide was the strongest Y4R PAM, significantly increasing IP accumulation following stimulation of Y4R with 1 nM PP (185.7 ± 23.5% (SEM) versus 100.00 ± 13.9%, p<0.05, n = 2; see Fig 1). Furthermore, Niclosamide had minor effects on Y1R (125.4 ± 14.6% versus 100.0 ± 28.3%, p>0.05, n = 2), Y2R (88.5 ± 6.2% versus 100.0 ± 14.9%, p>0.05, n = 2), and Y5R (83.4 ± 5.9% versus 100.0 ± 27.3% p>0.05, n = 2) when stimulated with 1 nM NPY. In addition to Niclosamide, adenosine and VU0244224 induced slight increases in the Y4R signal response in the IP3 assay (120 ± 11% and 127 ± 19%, respectively). Whereas VU0244224 had no effects on other Y receptor subtypes, adenosine appeared to slightly decrease signal transduction at Y1R and Y5R. These results show Niclosamide to be the most effective Y4R PAM hit compound with validated activity in a complementary assay and it alone was selected for further investigation (S1 Fig). Due to the lack of any viable hits with the exception of Niclosamide, a second screen was performed testing a total of 33,288 compounds. Of these 33,288 compounds, 32,000 were randomly selected from the VICB compound library. Since Niclosamide showed the strongest effect as a Y4R PAM, the collection of compounds for the second screening was enriched with 1,288 compounds structurally similar to Niclosamide based on Tanimoto coefficients (S1 Table). All potential Y4R PAMs were retested for nonspecific effects in wildtype COS-7 cells. Hit compounds that did not show any activity in wildtype COS-7 cells were tested on the Y4R in a concentration dependent manner for their effect on a PP EC20 and EC80. Validated hits were then tested for their effects on histamine and bradykinin receptor-evoked changes in intracellular Ca2+ in order to further exclude off-target effects that might, for instance, result from changes in common effectors downstream of the GPCR. In the second HTS, four compounds structurally similar to Niclosamide were identified as Y4R PAMs. Of these similar compounds, stability of VU0118748 was examined to ensure that activity was not due to hydrolysis yielding Niclosamide. HPLC analysis at different incubation times suggest that this compound is stable (S3 Fig). Along with two structurally related but inactive compounds of the VU compound library (VU0114795 and VU0357475), selected to supplement structure-activity relationship studies with YR selectivity, these compounds were further investigated for Y4R PAM activity and YR subtype selectivity (Fig 2).
Fig 2. Structures of Y4R PAMs identified by HTS and inactive control compounds chosen for further characterization of Y4R PAM activity and YR subtype selectivity.

Validation and selectivity of Y4R PAMs
After identification of Niclosamide-like compounds in the HTS, the Y4R PAM activity was again validated using an IP3 accumulation assay system. Compounds were investigated for potentiation of a PP EC20 in a concentration-dependent manner to determine their potency on the Y4R (Fig 3).
Fig 3. Y4R PAM activity of Niclosamide-like compounds.

Potency of the Y4R PAMs was investigated with an inositol phosphate accumulation assay through potentiation of a PP EC20 response. Data have been normalized to the maximum IP accumulation caused by the Y4R native ligand PP. Data represent the mean ± SEM of three independent experiments performed in duplicate.
Niclosamide, VU0048913, VU0118748, VU0048992 and VU0049150, identified in the Ca2+ flux HTS, potentiated the PP signal response in the IP3 accumulation assay. In presence of 30 μM of the active compounds, the PP response increased to approximately 40% (Fig 3) compared to the PP-evoked signal in the absence of the compounds. However, modifications on the Niclosamide scaffold affected the potency of the compounds. Niclosamide, VU0048913, and VU0118748 potentiated the PP EC20 with comparable EC50 values of 620 nM, 566 nM and 473 nM, respectively. In contrast, structural modifications in compound VU0048992 lead to a dramatic loss of Y4R potency (EC50 >10 μM) or completely inactive structures in case of VU0114795 and VU0357475.
To investigate the Y receptor subtype selectivity, we tested the effect of the Niclosamide-like structures (Fig 2) at all four human Y receptors with their native ligands (PP for Y4R and NPY for Y1R, Y2R, and Y5R). Full concentration-response relationships were determined for each ligand-receptor pair in the presence of DMSO vs. 30 μM compound (S2 Fig), the concentration at which all compounds had a comparable effect on the Y4R (Fig 3). This high compound concentration was used for response curves in S2 Fig due to failure of some compounds to reach maximal response in concentrations used in Fig 3. None of the tested compounds had an effect on the basal level or maximum level of the signal response (S2 Fig). Thus, the influence of the compounds on the agonists EC50 (pEC50 ± SEM) of the signal response was used as an indicator for selectivity (Fig 4).
Fig 4. YR subtype selectivity of Y4R PAMs.

Effect of 30 μM compound on the pEC50 of Y-receptor agonists in COS-7 cells stable expressing a Y receptor subtype and the chimeric G-protein Gα6qi4myr. Receptors were stimulated with their native ligands (Y1R, Y2R, Y5R: NPY; Y4R: PP). For Y-axis values, positive modulation represents an increase in the apparent potency (pEC50) of the native agonist and negative modulation represents a decrease in the apparent potency of the native agonist. Data represent the mean ± SEM of at least two independent experiments (for full concentration-response curves see S2 Fig) (*p < .05, ***p < .001 Bonferroni).
Niclosamide, VU0048913, VU0048992, VU0049150, and VU0118748 increase the potency of PP at the Y4R (Fig 4, S2 Fig) consistent with Y4R PAM activity observed (Fig 3). Testing of other Y receptor subtypes revealed that the compounds are not fully selective for the Y4R subtype. In addition to the Y4R activity, Niclosamide had a small PAM effect on the Y1R. However, the structural analogs VU0048913, VU0048992, VU0049150, and VU0118748 had no effect on the Y1R signal. In contrast to the PAM effects observed on Y4R, Niclosamide and VU0118748 show a negative allosteric effect on Y5R. All other tested compounds were inactive on the Y5R. Whereas Niclosamide had no effect on the Y2R, compounds VU0048913, VU0048992, and VU0114795 showed a slight PAM effect on the Y2R. Overall, the effects of the Y4R PAM compounds on Y1R, Y2R, and Y5R were lower than the PAM effect at the Y4R.
Niclosamide structure-activity relationships
Potentiation of PP EC20 experiments showed that Niclosamide, VU0048913, and VU0118748 have nearly identical potencies at the Y4R. Accordingly, the loss of the Cl atom (VU0048913) on the aniline ring structure, as well as modification of the OH group in the benzoyl ring (VU0118748) do not affect Y4R PAM activity. In contrast, a major change of the substituents meta or para to the hydroxyl function on the benzoyl ring led to a complete loss of Y4R PAM activity (VU0114795 and VU0357475) or drastically reduced Y4R potency (VU0048992 and VU0049150). As shown by the active compound VU0048913, the Cl on the benzoyl ring structure can be substituted by Br, which underlines the importance of the electron-rich character in this position for the potency to the Y4R (Figs 3 and 5).
Fig 5. Distinct positions of the Niclosamide scaffold were shown to be relevant for Y4R PAM activity and YR selectivity.

Substitutions in the benzoyl ring are important for Y4R potency (green), and offer a potential modification site (grey). Modifications in the aniline ring engender selectivity towards Y1R / Y5R subtype (red).
Furthermore, investigation of PAM activity on Y1R, Y2R and Y5R suggested potential modification sites to control YR selectivity. Removal of the Cl substitution on the aniline ring in VU0048913 reduced Y1R effects and Y5R antagonism. As shown by VU0118748, selectivity towards Y1R can also be achieved by the introduction of the nitrobenzoic-acid on the OH position in the benzoyl ring. However, this modification had no effect on the Y5R NAM activity and Y4R PAM activity, suggesting this position as a potential site to control subtype selectivity.
Discussion
Therapeutic potential
The application of allosteric modulators presents a promising approach for the treatment of complex receptor-ligand systems that regulate sensitive physiological processes such as nervous signal transduction or metabolic regulation [21, 30]. The benefits of Y4R modulation on obesity and insulin resistance are becoming more important as the number of patients diagnosed with type 2 diabetes rises alongside risk factors such as the prevalence of obesity, physical inactivity, and poor diet [31]. In humans, low circulating PP levels were found in obese children and adults [32]. Hyperphagia in obese patients can be reduced by restoring basal and meal-stimulated PP levels through IV infusion [33]. Additionally, PP is hypothesized to sensitize the liver to insulin through upregulation of the insulin receptor β subunit [34, 35]. In patients with diabetes secondary to chronic pancreatitis, PP administration reduces insulin resistance and improves glucose metabolism [36]. Effects of PP administration and the complex interplay of obesity and diabetes suggest Y4R modulation may be beneficial for a wide range of metabolic disorders. This is already being seen in preclinical studies of TM-30339, a PP based Y4R-selective peptide agonist and in phase I and II trials of Obineptide, an Y2/4R dual peptide agonist [37]. With respect to these diverse effects of the PP dependent Y4R activation, the availability of small molecule ligands and modulators offer a new possibility to get further insights into the pharmacology of this GPCR.
In this study, we present the first small molecule Y4R PAM. Niclosamide was identified as a Y4R PAM in the primary screen of the Spectrum Collection using the Ca2+ flux assay and its activity was validated in the alternative IP accumulation assay. The second HTS experiment identified four additional Y4R PAMs that are structurally similar to Niclosamide, confirming the importance of this scaffold for Y4R potentiation. YR subtype selectivity was characterized for four Niclosamide-like Y4R PAMs along with two Y4R inactive Niclosamide-derived structures.
Y4R potency and selectivity
Investigation of the potency of the compounds on the Y4R showed that three compounds, Niclosamide, VU0048913, and VU0118748, had comparable EC50 values of around 500 nM. All other compounds lacking the halogen substitution on the benzoyl ring were either completely inactive at the Y4R or had an at least 10-fold lower EC50 for potentiation of a PP EC20 (Fig 3). These investigations highlight the role of an electron-rich substituent at this position for Y4R potency. In contrast, the bulky nitro-benzoyl substitution in VU0118748 had no influence on Y4R potency and Y4R PAM activity. However, this modification lowers the effect at Y1R. This offers a role for this position as a potential modification site for improving Y4R selectivity while maintaining the Y4R PAM activity.
Selectivity studies of Niclosamide and analogs on all four Y receptor subtypes revealed that the compounds are not fully selective for the Y4R. NPY was used as the ligand for selectivity studies on Y1R, Y2R and Y5R, as it activates all three receptors with high potencies. However, allosteric modulators are known for the potential of probe dependence effects for different ligands of one GPCR and thus effects for alternative NPY ligands PYY or PP on these Y receptor subtypes cannot be excluded at this stage of the investigations. Niclosamide and VU0118748, both Y4R PAMs, showed antagonistic effects on the Y5R. The Y4R and Y5R fulfill different actions in the regulation of appetite and food intake. While the Y4R has an anorexigenic effect by inducing satiety in response to the activation of the native ligand PP, the Y1R and Y5R are characterized to have an orexigenic effect [38]. A simultaneous Y5R antagonism and Y4R PAM activity thus could contribute to an anti-obesity effect of Niclosamide or VU0118748. However, the different analogs of the compound Niclosamide scaffold suggest the possibility of developing Y4R PAMs with a higher degree of specificity relative to other Y receptor subtypes (Fig 5). It is not uncommon for compounds to produce a variety of effects at a common binding site. For example, recently a known metabotropic glutamate receptor 4 (mGluR4) PAM/mGluR1 NAM chemotype was converted into a selective mGluR1 PAMs by virtue of a double “molecular switch” [39].
Would an EC50 shift of 4 fold be sufficient to cause an in vivo effect?
Niclosamide and some related analogs induced a 4-fold increase in the potency of PP at Y4R. Allosteric modulators of other class A GPCRS, especially of neurotransmitter receptors, display a stronger allosteric effects with EC50 shifts >10 fold, shown for muscarinic receptor 4 (mAChR4) [40]. However, allosteric modulation of the CaSR by cinacalcet shows that even smaller in vitro effects can be effective in vivo and that clinical efficacy is dependent on the receptor, tissue and the metabolic state that is targeted [41]. This suggests that the comparatively small increase in PP potency caused by Niclosamide may be sufficient to elicit in vivo effects.
Niclosamide improves diabetic symptoms in mice
Niclosamide is an FDA approved anthelmintic drug that treats parasitic worm infection through the uncoupling of mitochondria. Interestingly, this compound was recently studied as a potential therapeutic for treatment of type 2 diabetes due to its high tolerability and the benefits of lipid mitochondrial uncoupling for treating diabetes [42]. Tao et al. fed mice the ethanolamine salt form of Niclosamide and showed it to be efficacious at high nanomolar concentrations (measured with blood sample liquid chromatography–tandem mass spectrometry at various time points) in reducing plasma insulin decline in db/db mice, sensitizing the insulin response, and preventing and treating diabetic symptoms during high fat diet induced obesity in mice. The authors focus on mitochondrial uncoupling as the primary mechanism of action for Niclosamide in the treatment of diabetes symptoms. However, our identification of Niclosamide as a Y4R PAM suggests that the efficacy of Niclosamide on diabetic symptoms may instead result from its action on the YR signalling in addition to effects on mitochondrial function.
Supporting Information
Structurally different small molecules (A) showed a positive effect on Y4R Ca2+ signal response in an HTS screening of the spectrum collection. Retesting in the IP3 assay as an alternative YR activation readout validated Niclosamide as a Y4R PAM (B) and offered other hits to have additional effects on other YR subtypes. Submaximal activation of Y receptors was observed for stimulation with 1 nM ligand (Y4R: PP, Y1,2,5R: NPY) in presence of 10 µM compound. Data represent the mean ± SEM of two independent experiments performed in quadruplicates (***p < .005 Bonferroni).
(TIF)
Receptor activation was investigated with an inositol phosphate accumulation assay in COS-7 cells stably expressing a Y receptor subtype and chimeric G-protein ΔGα6qi4myr. Data represent the mean ± SEM of at least 2 independent experiments, each performed in triplicate.
(TIF)
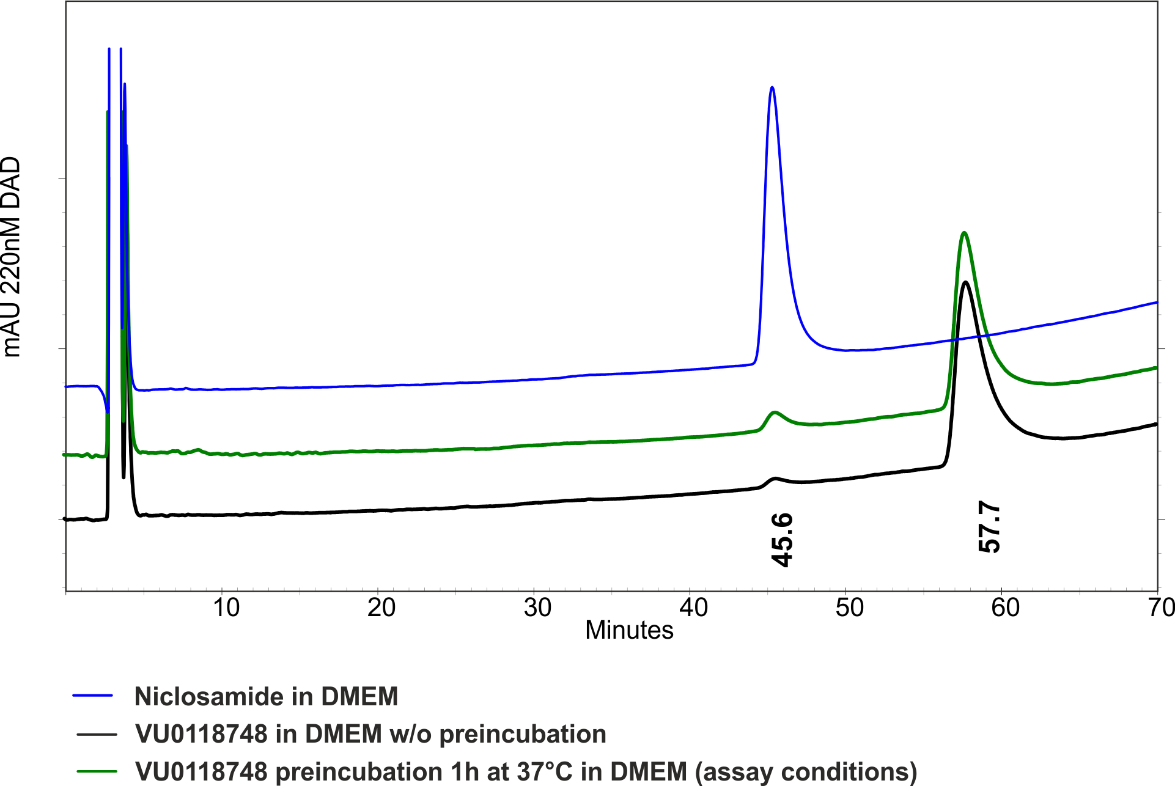
Niclosamide and VU0118748 showed different retention times of 46 min (63% ACN) and 58 min (71% ACN), respectively. VU0118748 in DMEM (without pre-incubation, black) shows a major signal with 98% integrated absorption at 58 min retention time, indicating the intact VU0118748. To test the stability of VU0118748 in the assay conditions, the compound was pre-incubated in DMEM for 2 hours at 37°C. HPLC analysis afterwards (green) shows a slightly increased fraction at 46 min retention time (5% of total absorption). Results show 95% of the compound is still intact.
(PNG)
{kind=link}
(PDF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was supported by National Institutes of Health [R01 DK097376 to JM], by the Deutsche Forschungsgemeinschaft (DFG) [Grant Be1264-16 to ABS], and by the European Union and the Free State of Saxony [Grants 100148835 and 1423213128452 to ABS]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health affairs. 2009;28(5):w822–31. Epub 2009/07/29. 10.1377/hlthaff.28.5.w822 . [DOI] [PubMed] [Google Scholar]
- 2.Troke RC, Tan TM, Bloom SR. The future role of gut hormones in the treatment of obesity. Therapeutic advances in chronic disease. 2014;5(1):4–14. Epub 2014/01/02. 10.1177/2040622313506730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makris A, Foster GD. Dietary approaches to the treatment of obesity. The Psychiatric clinics of North America. 2011;34(4):813–27. Epub 2011/11/22. 10.1016/j.psc.2011.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kissler HJ, Settmacher U. Bariatric surgery to treat obesity. Seminars in nephrology. 2013;33(1):75–89. Epub 2013/02/05. 10.1016/j.semnephrol.2012.12.004 . [DOI] [PubMed] [Google Scholar]
- 5.Smith BR, Schauer P, Nguyen NT. Surgical approaches to the treatment of obesity: bariatric surgery. Endocrinology and metabolism clinics of North America. 2008;37(4):943–64. Epub 2008/11/26. 10.1016/j.ecl.2008.08.001 . [DOI] [PubMed] [Google Scholar]
- 6.Heppner KM, Perez-Tilve D. GLP-1 based therapeutics: simultaneously combating T2DM and obesity. Frontiers in neuroscience. 2015;9:92 Epub 2015/04/09. 10.3389/fnins.2015.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Field BC, Chaudhri OB, Bloom SR . Bowels control brain: gut hormones and obesity. Nature reviews Endocrinology. 2010;6(8):444–53. Epub 2010/06/30. 10.1038/nrendo.2010.93 . [DOI] [PubMed] [Google Scholar]
- 8.Pedragosa-Badia X, Stichel J, Beck-Sickinger AG. Neuropeptide Y receptors: how to get subtype selectivity. Frontiers in endocrinology. 2013;4:5 Epub 2013/02/06. 10.3389/fendo.2013.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asakawa A, Inui A, Yuzuriha H, Ueno N, Katsuura G, Fujimiya M, et al. Characterization of the effects of pancreatic polypeptide in the regulation of energy balance. Gastroenterology. 2003;124(5):1325–36. Epub 2003/05/06. . [DOI] [PubMed] [Google Scholar]
- 10.Jesudason DR, Monteiro MP, McGowan BMC, Neary NM, Park AJ, Philippou E, et al. Low-dose pancreatic polypeptide inhibits food intake in man. British Journal of Nutrition. 2007;97(03):426–9. 10.1017/S0007114507336799 [DOI] [PubMed] [Google Scholar]
- 11.le Roux CW, Batterham RL, Aylwin SJ, Patterson M, Borg CM, Wynne KJ, et al. Attenuated peptide YY release in obese subjects is associated with reduced satiety. Endocrinology. 2006;147(1):3–8. Epub 2005/09/17. 10.1210/en.2005-0972 . [DOI] [PubMed] [Google Scholar]
- 12.Ekblad E, Sundler F. Distribution of pancreatic polypeptide and peptide YY. Peptides. 2002;23(2):251–61. Epub 2002/02/05. . [DOI] [PubMed] [Google Scholar]
- 13.Cox HM. Neuropeptide Y receptors; antisecretory control of intestinal epithelial function. Autonomic neuroscience: basic & clinical. 2007;133(1):76–85. Epub 2006/12/05. 10.1016/j.autneu.2006.10.005 . [DOI] [PubMed] [Google Scholar]
- 14.Kojima S, Ueno N, Asakawa A, Sagiyama K, Naruo T, Mizuno S, et al. A role for pancreatic polypeptide in feeding and body weight regulation. Peptides. 2007;28(2):459–63. Epub 2007/01/09. 10.1016/j.peptides.2006.09.024 . [DOI] [PubMed] [Google Scholar]
- 15.Holzer P, Reichmann F, Farzi A. Neuropeptide Y, peptide YY and pancreatic polypeptide in the gut–brain axis. Neuropeptides. 2012;46(6):261–74. 10.1016/j.npep.2012.08.005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katsuura G, Asakawa A, Inui A. Roles of pancreatic polypeptide in regulation of food intake. Peptides. 2002;23(2):323–9. Epub 2002/02/05. . [DOI] [PubMed] [Google Scholar]
- 17.Acuna-Goycolea C, Tamamaki N, Yanagawa Y, Obata K, van den Pol AN. Mechanisms of Neuropeptide Y, Peptide YY, and Pancreatic Polypeptide Inhibition of Identified Green Fluorescent Protein-Expressing GABA Neurons in the Hypothalamic Neuroendocrine Arcuate Nucleus. The Journal of Neuroscience. 2005;25(32):7406–19. 10.1523/jneurosci.1008-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asakawa A, Inui A, Ueno N, Fujimiya M, Fujino MA, Kasuga M. Mouse pancreatic polypeptide modulates food intake, while not influencing anxiety in mice. Peptides. 1999;20(12):1445–8. Epub 2000/03/04. . [DOI] [PubMed] [Google Scholar]
- 19.Misra S, Murthy KS, Zhou H, Grider JR. Coexpression of Y1, Y2, and Y4 receptors in smooth muscle coupled to distinct signaling pathways. The Journal of pharmacology and experimental therapeutics. 2004;311(3):1154–62. Epub 2004/08/17. 10.1124/jpet.104.071415 . [DOI] [PubMed] [Google Scholar]
- 20.Ueno N, Inui A, Iwamoto M, Kaga T, Asakawa A, Okita M, et al. Decreased food intake and body weight in pancreatic polypeptide-overexpressing mice. Gastroenterology. 1999;117(6):1427–32. Epub 1999/12/02. . [DOI] [PubMed] [Google Scholar]
- 21.Conn PJ, Lindsley CW, Meiler J, Niswender CM. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nature reviews Drug discovery. 2014;13(9):692–708. 10.1038/nrd4308 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shonberg J, Lopez L, Scammells PJ, Christopoulos A, Capuano B, Lane JR. Biased agonism at G protein-coupled receptors: the promise and the challenges—a medicinal chemistry perspective. Medicinal research reviews. 2014;34(6):1286–330. Epub 2014/05/07. 10.1002/med.21318 . [DOI] [PubMed] [Google Scholar]
- 23.Keov P, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors: A pharmacological perspective. Neuropharmacology. 2011;60(1):24–35. 10.1016/j.neuropharm.2010.07.010. 10.1016/j.neuropharm.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 24.Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. British Journal of Pharmacology. 2012;165(6):1659–69. 10.1111/j.1476-5381.2011.01749.x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregory KJ, Dong EN, Meiler J, Conn PJ. Allosteric Modulation of Metabotropic Glutamate Receptors: Structural Insights and Therapeutic Potential. Neuropharmacology. 2011;60(1):66–81. 10.1016/j.neuropharm.2010.07.007 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang R, Xie X. Tools for GPCR drug discovery. Acta pharmacologica Sinica. 2012;33(3):372–84. Epub 2012/01/24. 10.1038/aps.2011.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mäde V, Bellmann-Sickert K, Kaiser A, Meiler J, Beck-Sickinger AG. Position and Length of Fatty Acids Strongly Affect Receptor Selectivity Pattern of Human Pancreatic Polypeptide Analogues. ChemMedChem. 2014;9(11):2463–74. 10.1002/cmdc.201402235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaiser A, Müller P, Zellmann T, Scheidt HA, Thomas L, Bosse M, et al. Unwinding of the C-Terminal Residues of Neuropeptide Y is critical for Y2 Receptor Binding and Activation. Angewandte Chemie International Edition. 2015;54(25):7446–9. 10.1002/anie.201411688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedragosa-Badia X, Sliwoski GR, Dong Nguyen E, Lindner D, Stichel J, Kaufmann KW, et al. Pancreatic polypeptide is recognized by two hydrophobic domains of the human Y4 receptor binding pocket. The Journal of biological chemistry. 2014;289(9):5846–59. Epub 2014/01/01. 10.1074/jbc.M113.502021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Martin B, Brenneman R, Luttrell LM, Maudsley S. Allosteric modulators of g protein-coupled receptors: future therapeutics for complex physiological disorders. The Journal of pharmacology and experimental therapeutics. 2009;331(2):340–8. Epub 2009/08/12. 10.1124/jpet.109.156380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ershow AG. Environmental influences on development of type 2 diabetes and obesity: challenges in personalizing prevention and management. Journal of diabetes science and technology. 2009;3(4):727–34. Epub 2010/02/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yulyaningsih E, Zhang L, Herzog H, Sainsbury A. NPY receptors as potential targets for anti-obesity drug development. Br J Pharmacol. 2011;163(6):1170–202. Epub 2011/05/07. 10.1111/j.1476-5381.2011.01363.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berntson GG, Zipf WB, O'Dorisio TM, Hoffman JA, Chance RE. Pancreatic polypeptide infusions reduce food intake in Prader-Willi syndrome. Peptides. 1993;14(3):497–503. Epub 1993/05/01. . [DOI] [PubMed] [Google Scholar]
- 34.Seymour NE, Volpert AR, Andersen DK. Regulation of hepatic insulin receptors by pancreatic polypeptide in fasting and feeding. The Journal of surgical research. 1996;65(1):1–4. Epub 1996/09/01. 10.1006/jsre.1996.9999 . [DOI] [PubMed] [Google Scholar]
- 35.Inui A. Transgenic approach to the study of body weight regulation. Pharmacological reviews. 2000;52(1):35–61. Epub 2000/03/04. . [PubMed] [Google Scholar]
- 36.Brunicardi FC, Chaiken RL, Ryan AS, Seymour NE, Hoffmann JA, Lebovitz HE, et al. Pancreatic polypeptide administration improves abnormal glucose metabolism in patients with chronic pancreatitis. The Journal of clinical endocrinology and metabolism. 1996;81(10):3566–72. Epub 1996/10/01. 10.1210/jcem.81.10.8855802 . [DOI] [PubMed] [Google Scholar]
- 37.Sato N, Ogino Y, Mashiko S, Ando M. Modulation of neuropeptide Y receptors for the treatment of obesity. Expert Opin Ther Pat. 2009;19(10):1401–15. Epub 2009/09/12. 10.1517/13543770903251722 . [DOI] [PubMed] [Google Scholar]
- 38.Moreno-Herrera A, Garcia A, Palos I, Rivera G. Neuropeptide Y1 and Y5 Receptor Antagonists as Potential Anti-Obesity Drugs. Current Status. Mini reviews in medicinal chemistry. 2014. Epub 2014/10/31. . [PubMed] [Google Scholar]
- 39.Cho HP, Garcia-Barrantes PM, Brogan JT, Hopkins CR, Niswender CM, Rodriguez AL, et al. Chemical modulation of mutant mGlu1 receptors derived from deleterious GRM1 mutations found in schizophrenics. Acs Chem Biol. 2014;9(10):2334–46. Epub 2014/08/20. 10.1021/cb500560h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, et al. Allosteric modulation of the muscarinic M(4) receptor as an approach to treating schizophrenia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(31):10978–83. 10.1073/pnas.0800567105 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A. Positive and Negative Allosteric Modulators Promote Biased Signaling at the Calcium-Sensing Receptor. Endocrinology. 2011;153(3):1232–41. 10.1210/en.2011-1426 [DOI] [PubMed] [Google Scholar]
- 42.Tao H, Zhang Y, Zeng X, Shulman GI, Jin S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nature medicine. 2014;20(11):1263–9. Epub 2014/10/06. 10.1038/nm.3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structurally different small molecules (A) showed a positive effect on Y4R Ca2+ signal response in an HTS screening of the spectrum collection. Retesting in the IP3 assay as an alternative YR activation readout validated Niclosamide as a Y4R PAM (B) and offered other hits to have additional effects on other YR subtypes. Submaximal activation of Y receptors was observed for stimulation with 1 nM ligand (Y4R: PP, Y1,2,5R: NPY) in presence of 10 µM compound. Data represent the mean ± SEM of two independent experiments performed in quadruplicates (***p < .005 Bonferroni).
(TIF)
Receptor activation was investigated with an inositol phosphate accumulation assay in COS-7 cells stably expressing a Y receptor subtype and chimeric G-protein ΔGα6qi4myr. Data represent the mean ± SEM of at least 2 independent experiments, each performed in triplicate.
(TIF)
Niclosamide and VU0118748 showed different retention times of 46 min (63% ACN) and 58 min (71% ACN), respectively. VU0118748 in DMEM (without pre-incubation, black) shows a major signal with 98% integrated absorption at 58 min retention time, indicating the intact VU0118748. To test the stability of VU0118748 in the assay conditions, the compound was pre-incubated in DMEM for 2 hours at 37°C. HPLC analysis afterwards (green) shows a slightly increased fraction at 46 min retention time (5% of total absorption). Results show 95% of the compound is still intact.
(PNG)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.