

Abstract
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are the clinical manifestations of severe lung damage and respiratory failure. Characterized by severe inflammation and compromised lung function, ALI/ARDS result in very high mortality of affected individuals. Currently, there are no effective treatments for ALI/ARDS, and ironically, therapies intended to aid patients (specifically mechanical ventilation, MV) may aggravate the symptoms. Key events contributing to the development of ALI/ARDS are: increased oxidative and proteotoxic stresses, unresolved inflammation, and compromised alveolar-capillary barrier function. Since the airways and lung tissues are constantly exposed to gaseous oxygen and airborne toxicants, the bronchial and alveolar epithelial cells are under higher oxidative stress than other tissues. Cellular protection against oxidative stress and xenobiotics is mainly conferred by Nrf2, a transcription factor that promotes the expression of genes that regulate oxidative stress, xenobiotic metabolism and excretion, inflammation, apoptosis, autophagy, and cellular bioenergetics. Numerous studies have demonstrated the importance of Nrf2 activation in the protection against ALI/ARDS, as pharmacological activation of Nrf2 prevents the occurrence or mitigates the severity of ALI/ARDS. Another promising new therapeutic strategy in the prevention and treatment of ALI/ARDS is the activation of autophagy, a bulk protein and organelle degradation pathway. In this review, we will discuss the strategy of concerted activation of Nrf2 and autophagy as a preventive and therapeutic intervention to ameliorate ALI/ARDS.
Keywords: Acute lung injury, oxidative stress, Nrf2, autophagy
Acute Lung Injury and Acute Respiratory Distress Syndrome
Acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS), are severe inflammatory diseases of the lung caused by disruption of the lung endothelial (capillaries) and epithelial (alveoli) barriers. ARDS was first described by Ashbaugh et al. in 1967, but it was not until 1994 that a consensus definition and description of ALI/ARDS was achieved by the American-European Consensus Conference (AECC) (Ashbaugh et al. 1967). The clinical criteria that define ALI/ARDS are: 1) acute onset of respiratory symptoms, 2) chest radiographs with bilateral infiltrates, 3) a ratio of partial pressure of oxygen in arterial blood (PaO2) to fraction of inspired oxygen (FiO2) of <200 mmHg for ARDS and <300 mmHg for ALI, 4) pulmonary capillary wedge pressure (PCWP) of less than 18 mmHg, and 5) the absence of clinical evidence of primary left atrial hypertension (Bernard et al. 1994).
It is estimated that 64.2–78.9 per 100,000 persons per year develop ALI, which has a mortality rate of 38.5% (Walkey et al. 2012). Based on these data, approximately 190,600 people in the United States each year will develop ALI, and 74,500 people will not survive (Rubenfeld et al. 2005; Walkey et al. 2012). Some survivors recover completely (Walkey et al. 2012; Herridge et al. 2011); however, others may have lasting damage to their lungs and develop additional health problems (Walkey et al. 2012; Mamary et al. 2011).
Pathogenesis of ALI/ARDS
As ALI/ARDS is a syndrome, or a collection of signs and symptoms, and not a specific disease, there are many insults that can produce lung injury directly or indirectly and cause ALI/ARDS (Bernard et al. 1994). Direct pulmonary injury involves a disease process that begins in the lungs by causing primarily lung damage, such as lung contusions, diffuse pulmonary infections (bacterial, viral, and fungal pneumonia), aspiration of gastric contents, near drowning, inhalation of toxic substances, or hyperoxia (Bernard et al. 1994). ALI/ARDS can be induced or aggravated by mechanical ventilation (MV), a therapeutic intervention used to assist patients with compromised respiratory functions due to edema, trauma, or general anesthesia (Wright 2014). MV causes ventilation-induced lung injury (VILI) due to different types of lung injury, including: 1) volutrauma, from lung over-distension; 2) barotrauma, from the direct effect of high pressure on the lung; 3) atelectrauma, from the shear stress of repetitive opening and closing of alveoli; and 4) biotrauma, from the generation of cytokines and inflammatory cascades (Slutsky 2005). Indirect pulmonary injury is a systemic disease process that affects the lungs secondarily, as in sepsis syndrome and systemic inflammatory response syndrome (SIRS), severe non-thoracic trauma, shock, acute pancreatitis, cardiopulmonary bypass, transfusion-related ALI (TRALI), disseminated intravascular coagulation (DIC), and burns (Bernard et al. 1994).
Direct or indirect injury to the lung parenchyma leads to damage of type I and type II epithelial cells of the alveoli, and of endothelial cells of the pulmonary capillaries, resulting in loss of the alveolar-capillary barrier (Beasley 2010). Without this barrier, fluid from the capillaries leaks into the interstitium and the alveoli, causing pulmonary edema, collapse of the lungs (atelectasis), and respiratory failure (Beasley 2010). These classic histological manifestations of ARDS in the lung are called diffuse alveolar damage (DAD) (Beasley 2010). The pathogenesis of DAD begins with an acute/exudative stage (≤7 days after insult), characterized by damage to the alveolar-capillary barrier, resulting in the formation of an exudate in alveolar spaces and interstitium, activation of resident alveolar macrophages, and release of pro-inflammatory cytokines to promote neutrophil infiltration. Macrophages and neutrophils secrete various proteases and oxidants that further injure alveolar endothelial and epithelial cells and digest the alveolar stroma. As a result, type II alveolar epithelial cells and fibroblasts proliferate in an attempt to repair the lung injury during the organizing/fibroproliferative stage (Beasley 2010). DAD may gradually resolve within 6–12 months; however, the most common outcome is a fibrotic stage that results in chronic respiratory compromise (Beasley 2010; Matthay et al. 2012).
Experimental models for studying ALI/ARDS in vivo
Based on the diverse etiology of ALI/ARDS, many different modeling strategies have been developed in animals in an attempt to reproduce the features of human ALI/ARDS. Direct lung injury in animals can be induced by 1) the intratracheal or intranasal administration of bacteria or bacterial products, such as lipopolysaccharide (LPS) from the outer cell wall of gram negative bacteria, or peptidoglycan and lipoteichoic acid from gram positive bacteria, to reproduce diffuse pneumonia; 2) the administration of hydrochloric acid or gastric particulates to reproduce aspiration; 3) the administration of high inspired fractions of oxygen to induce hyperoxia; or 4) the induction of ischemia/reperfusion by clamping the hilum (Matute-Bello et al. 2008). Indirect lung injury is based on 1) reproducing sepsis using cecal ligation and puncture, 2) the administration of intravenous bacteria or LPS, or 3) mesenteric ischemia/reperfusion (Matute-Bello et al. 2008). Animal models of VILI consist of mechanical ventilation with high tidal volumes and low positive end-expiratory pressure (Wilson, Takata 2013).
Current Therapeutic and Treatment Options for ALI/ARDS
The treatment for ARDS is mainly supportive and focuses on maintaining ventilation and oxygenation, normal cardiac function, and nutritional support, as well as the administration of nitric oxide (NO) and corticosteroids, and the prevention of further complications (Ware, Matthay 2000; Matthay et al. 2012). A more appealing strategy would be to treat the underlying cause for the lung injury and respiratory distress (Ware, Matthay 2000; Matthay et al. 2012). Central to the pathogenesis of ALI/ARDS is the generation of reactive oxygen species (ROS) that cause oxidative stress. Multiple cell types in the lungs, including endothelial cells, neutrophils, eosinophils, alveolar macrophages, and alveolar epithelial cells, are major ROS generators (Mittal et al. 2014). ROS may be produced by inflammatory cells (macrophages and neutrophils) trying to fight infections, or by dysfunctional mitochondria or enzymatic systems in alveolar epithelial cells and capillary endothelial cells after lung tissue damage by hyperoxia, electrophilic xenobiotics, and shear stress (Brune et al. 2013; Warabi et al. 2007; Bernard et al. 2014; Reddy et al. 2009c). Antioxidants have been shown to reduce the severity of ALI/ARDS in multiple mouse models that use different initiating insults, highlighting the central role of ROS in the pathophysiology of ALI/ARDS (Aggarwal et al. 2012; Howard et al. 2014; Ye et al. 2015; Husari et al. 2014; Shohrati et al. 2014; Zhao et al. 2014; Lingaraju et al. 2015; Hu et al. 2015; Yilmaz et al. 2014; Yamamoto et al. 2012; Campos et al. 2012).
The Nrf2 signaling pathway
Nrf2 (nuclear factor erythroid-2 related factor 2) is a transcription factor that regulates an adaptive cellular defense response to various stresses, including oxidative, proteotoxic, and metabolic stresses, as well as inflammation (Figure 1). Nrf2 heterodimerizes with small Maf (sMaf) proteins and together they bind the antioxidant response elements (AREs) in the regulatory regions of Nrf2 target genes that participate in the regulation of oxidative stress (HO-1, GCLM, TXNRD1), xenobiotic metabolism and excretion (NQO1, AKR1C1, GSTA, MRP1), inflammation (TGF-β, NF-κB), apoptosis (Bcl-2, Bcl-xL), autophagy (p62), and cellular bioenergetics (G6PD, PPARγ) (Figure 1) (Dinkova-Kostova, Abramov 2015; Kensler et al. 2007; Jaramillo, Zhang 2013). Even though Nrf2 is ubiquitously expressed in all cell types and tissues, its protein levels are kept low under basal (homeostatic) conditions since it is constantly degraded. The main negative regulator of Nrf2 is Keap1 (Kelch-like ECH associated protein 1), a substrate adaptor for a Cullin 3 (Cul3)-containing E3 ubiquitin ligase complex that ubiquitinates Nrf2 to promote its degradation by the 26S proteasome (Zhang 2006; Villeneuve et al. 2010; Kobayashi et al. 2004). Nrf2 binds through its N-terminal ETGE and DLG motifs to two Keap1 proteins at the Kelch domain in what is known as the hinge-and-latch model: the ETGE (hinge) binds to the Kelch domain in Keap1 more strongly than the DLG (latch) motif, which oscillates between open and closed conformations (Tong et al. 2006; Tong et al. 2007; Baird et al. 2013). Nrf2 ubiquitination occurs only when both of its motifs are binding to Keap1 in a closed conformation (Baird et al. 2013). Keap1 is a sensor of the intracellular redox status: oxidants and electrophilic chemopreventive compounds covalently modify several important cysteine residues, in particular cysteine 151 (C151), in Keap1. Oxidation or adduction of C151 causes a conformational change in Keap1 that affects the DLG-Kelch interaction, thus preventing Nrf2 ubiquitination and subsequent degradation (Zhang et al. 2004; Zhang, Hannink 2003). Consequently, Nrf2 protein is stabilized, accumulates, and translocates into the nucleus to activate the transcription of its target genes. Many known electrophilic chemopreventive compounds activate Nrf2 signaling through the canonical mechanism, and mutation of cysteine 151 to serine (Keap1-C151S) in Keap1 completely abolishes Nrf2 upregulation by canonical Nrf2 activators (Jaramillo, Zhang 2013). Signal termination occurs when the redox balance is restored, or when the chemopreventive compounds are metabolized and eliminated. At this point, Keap1 shuttles into the nucleus, binds to Nrf2, and brings it back into the cytosol for degradation (Sun et al. 2011).
Fig. 1. Nrf2 and autophagy are activated during ALI/ARDS.
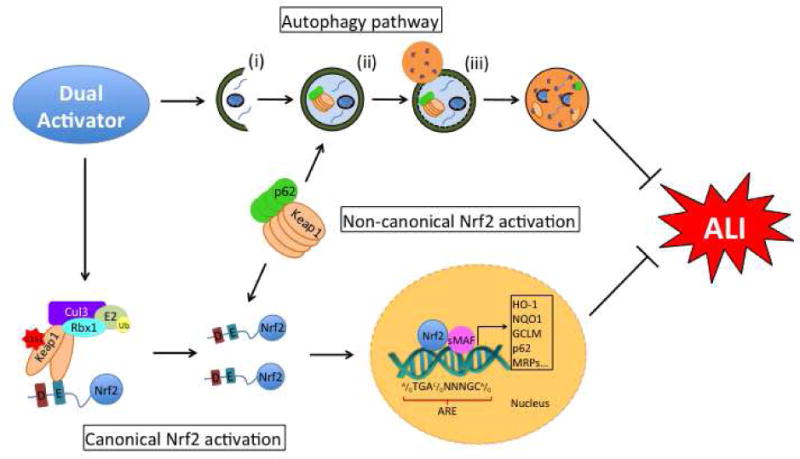
During ALI, production of reactive oxygen species activates Nrf2 and autophagy. Nrf2 pathway. The transcription factor Nrf2 binds to Keap1-Cul3-Rbx1 E3 ubiquitin ligase complex to be ubiquitinated and degraded. During ALI/ARDS, ROS modify cysteine 151 in Keap1 (C151), affecting its interaction with Nrf2. As a result, Nrf2 is no longer ubiquitinated and is stabilized. Newly synthesized Nrf2 accumulates in the cytosol, translocates to the nucleus, dimerizes with small Maf proteins, and binds to the antioxidant response element (ARE) in the regulatory regions of its target genes to promote their transcription (Canonical Nrf2 activation). Nrf2 can also be regulated by autophagy during ALI/ARDS. The autophagy substrate protein p62 binds to Keap1, sequestering it into the autophagosome and allowing for Nrf2 accumulation (Non-canonical Nrf2 activation). Autophagy pathway. ALI/ARDS induce autophagy, a bulk degradation pathway that occurs in three main stages: i) initiation, ii) elongation, and iii) fusion. Initiation involves the formation of the phagophore, which then elongates to encapsulate damaged proteins and organelles in a double membrane bound autophagosome. The autophagosome then fuses with the lysosome, and the damaged cargo is degraded by lysosomal hydrolases.
The p62-dependent, non-canonical mechanism, of Nrf2 regulation
Recent studies have identified other Nrf2 regulatory protein complexes that participate in other pathophysiological processes (Rada et al. 2011; Rada et al. 2012; Chowdhry et al. 2013; Wu et al. 2014). One of these corresponds to the autophagy-related protein p62 (sequestosome 1, SQSTM1), which activates Nrf2 (Lau et al. 2010). Autophagy is a bulk degradation pathway that involves the sequestration of damaged cellular components into an autophagosome, which fuses with the lysosome to form an autolysosome that degrades the cargo using lysosomal enzymes (see below for a more detailed description of the pathway and its role in ALI). p62 is an adaptor protein that recognizes the degradation cargo and brings it to the autophagosome; importantly, p62 is itself an autophagy degradation target. Autophagy inhibition at the late stage causes accumulation of autophagosomes and results in pathological activation of Nrf2 signaling (Lau et al. 2010; Jain et al. 2010; Komatsu et al. 2010; Riley et al. 2010; Fan et al. 2010). This occurs because p62 contains a pSTGE motif that competes with the ETGE/DLG motifs in Nrf2 for Keap1 binding (Jain et al. 2010; Ichimura et al. 2013). When the number of autophagosomes increases, p62 sequesters Keap1 and thus Nrf2 is stabilized (Figure 1) (Jiang et al. 2015). Our group called this mode of Nrf2 activation the non-canonical mechanism, as it is Keap1-C151-independent but p62-dependent (Lau et al. 2010). For example, arsenic, a human carcinogen, is an Nrf2 activator that induces Nrf2 through blockage of autophagy-lysosomal fusion (Lau et al. 2013). This mode of Nrf2 activation arises from a pathological state (autophagy dysregulation) and results in prolonged Nrf2 activation (the “dark side” of Nrf2); therefore, it is not desirable for cellular protection. On the other hand, induction of autophagy (increased autophagy flux) leads to the sequestration of Keap1-p62 complexes into autophagosomes, resulting in lysosomal-mediated degradation of Keap1 and controlled Nrf2 activation. This mode of Nrf2 activation is intermittent and confers protection. Nrf2 also promotes the ARE-driven expression of p62, indicating that autophagy and the Nrf2-mediated antioxidant response work in concert to restore cellular homeostasis (Taguchi et al. 2012).
The protective role of Nrf2 in ALI
The airway and lung epithelial cells constitute an important interphase between the body and the environment, and as such, these tissues are constantly exposed to oxidative stress and toxicants (Cho, Kleeberger 2014). Therefore, the expression of intra and extracellular antioxidants in airway epithelial cells and fluids is prominent. Numerous in vitro and in vivo studies have demonstrated the importance of Nrf2 activation to decrease oxidative stress and inflammation in conditions such as pulmonary fibrosis, cystic fibrosis, emphysema, COPD, ALI, asthma, bronchopulmonary dysplasia, and airway infections (Cho, Kleeberger 2015; Chan, Kan 1999). In the particular context of ALI/ARDS, the benefits of both basal and induced Nrf2 signaling have been investigated in various cellular and mouse models, as well as in human patient samples, as will be discussed below.
As mentioned above, the main regulation of Nrf2 occurs post-transcriptionally; however, over 500 single nucleotide polymorphisms (SNPs) in the regulatory and coding regions of Nrf2 have been identified (Cho 2013). The first evidence that Nrf2 had a role in ALI came from identification of Nrf2 as a candidate gene for ALI susceptibility by positional cloning in mice and subsequent analysis of SNPs in human populations and in vitro functional studies (Marzec et al. 2007). Multiple studies have determined that SNPs in Nrf2 confer susceptibility to ALI and other respiratory diseases like COPD and asthma (Cho et al. 2015a; Cordova et al. 2011; Cho et al. 2015c). In a hyperoxia-induced ALI mouse model, the functional effects of promoter and coding SNPs revealed that some haplotypes characteristic to certain inbred mouse strains confer increased susceptibility to hyperoxia due to lower Nrf2 mRNA expression and compromised protein function (Cho et al. 2015a). These studies will help identify populations at greater risk of developing ALI that would benefit from more effective chemopreventive interventions through Nrf2 upregulation.
Hyperoxia-induced ALI causes oxidative lung damage. Nrf2−/− mice were more susceptible to hyperoxia (95–98% oxygen, 72 h) and developed more severe ALI-like phenotypes than Nrf2+/+ mice, as determined by increased lung permeability, inflammation, and lung epithelial damage (Cho et al. 2002). In Nrf2−/− mice, both the basal and induced expression of Nrf2 target genes were lower than in Nrf2+/+, demonstrating that Nrf2 transcriptional activity is key in the response to hyperoxia-induced ALI. Furthermore, conditional deletion of Nrf2 in club cells (Clara cells, secretory bronchoepithelial cells) using cell type-specific Cre recombinase (CCSP-Cre × Nrf2fl/fl) yielded mice more sensitive to hyperoxia-induced ALI, confirming that Nrf2 activity in the airways is necessary for protection in this model system (Reddy et al. 2011). Additionally, unresolved oxidative stress and inflammation in Nrf2−/− mice increased apoptosis in sublethal hyperoxic exposure (48 h) and compromised tissue repair (Reddy et al. 2009a). These results indicate that Nrf2 has additional roles that go beyond the resolution of oxidative stress, including the regulation of inflammation and tissue remodeling factors (Cho, Kleeberger 2015). Currently, it is unknown if Nrf2 modulates these processes directly or indirectly, so further detailed mechanistic studies are needed to elucidate these Nrf2-dependent effects. Pharmacological Nrf2 activation as a strategy to prevent and treat hyperoxia-induced ALI has been explored. The Nrf2 activator CDDO-Im (a synthetic triterpenoid compound, CDDO-imidazole) conferred resistance against hyperoxia (Reddy et al. 2009b). In contrast to the clear effects of Nrf2 upregulation, administration of direct antioxidants like N-acetylcysteine (NAC), has limited efficacy in protection against hyperoxia-induced ALI. Administration of antioxidants to quench ROS is a strategy with limited effectiveness due to their limited availability and the fact that once oxidized they are useless, and some antioxidants only scavenge certain ROS selectively. However, Nrf2 activation promotes sustained expression of endogenous antioxidants, detoxifying/excretion enzymes, repair and degradation proteins, and metabolic reprogramming (Dinkova-Kostova, Abramov 2015). Together, these Nrf2 downstream effector proteins remove insults, neutralize ROS, and repair damage to restore cellular homeostasis. In addition, Nrf2 regulates the expression of anti-inflammatory, pro-proliferative, anti-apoptotic, and autophagy-related genes, constituting a more holistic approach to counteract and repair the damage (Jaramillo, Zhang 2013). As such, Nrf2 activation provides not only short-term beneficial effects but also confers medium and long-term protection.
In high tidal volume MV models that induce VILI/ALI, Nrf2−/− mice have greater structural damage, increased alveolar-capillary permeability, greater inflammation and oxidative stress than Nrf2+/+ mice (Papaiahgari et al. 2007). Supplementation of NAC decreased VILI in Nrf2−/− mice, indicating that oxidative stress is a major contributing factor in the pathogenesis of VILI. Subsequent studies have been performed to evaluate the effects of Nrf2 activation in the protection against VILI. Sodium sulfide protects against VILI by upregulating Nrf2 target genes involved in the restoration of redox balance (Francis et al. 2011). Our group recently demonstrated that a newly identified Nrf2 activator, the carotenoid and food additive bixin, protects against VILI only in Nrf2+/+ but not Nrf2−/− mice (Tao et al. 2015).
ALI-like symptoms can also be chemically induced in mice using agents that cause oxidative damage and inflammation. The phenolic antioxidant butylated hydroxytoluene (BHT) is used as food additive to prevent the oxidation of lipids, and while it is relatively safe in humans, it is toxic when administered to mice. Oral administration of BHT is noxious to alveolar type I epithelial cells, the cells that comprise most of the alveolar structure and are responsible for the gas exchange and barrier functions (Ward, Nicholas 1984). BHT causes ALI-like symptoms in the lungs of Nrf2+/+ and Nrf2−/− mice, which develop massive edema and hemorrhage; however, Nrf2−/− mice are much more susceptible (Chan, Kan 1999). The precise cytotoxic mechanism of BHT in the lung has not been clarified, but oxidative stress may play a central role as Nrf2+/+ mice are more resistant (Cho et al. 2006). The effects of oxidative lung damage have been studied in an ozone (O3) mouse model. O3 is a strong oxidant and air pollutant that greatly affects the airways and lungs. Predictably, Nrf2 deficiency in mice exacerbates oxidant-induced lung injury (Cho et al. 2013). An important implication of this study is that Nrf2 activation by chemopreventive compounds could be very beneficial for populations living in areas with poor air quality exposed to very high O3. Carrageenin, a seaweed-derived polysaccharide used as a thickening and emulsifying agent in food and pharmaceuticals, induces acute inflammation and ALI-like symptoms after intratracheal injection (Mochizuki et al. 2005). Consistently, Nrf2−/− mice are more susceptible to carrageenin-induced ALI than Nrf2+/+ mice (Mochizuki et al. 2005). This study also helped to elucidate the anti-inflammatory functions of Nrf2. Carrageenin-induced inflammation induced COX-2 (cyclooxygenase 2), an enzyme that converts arachidonic acid into prostaglandins, which are mediators of inflammation (Ricciotti, FitzGerald 2011). In particular, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) activates Nrf2, and pharmacological inhibition of COX-2 induced severe ALI in Nrf2+/+ mice comparable to that of Nrf2−/−, which may be due at least in part to decreased expression of Nrf2 target genes (Ricciotti, FitzGerald 2011; Mochizuki et al. 2005). These results indicate that resolution of inflammation and oxidative injury are both important to prevent ALI.
Extensive work has been performed to characterize the biological effects of acute exposure to certain gases that represent particularly important hazards, either for their use as chemical warfare agents or due to common work place exposure. These studies have demonstrated that lung epithelial cells are among the first to be exposed to toxic gases such as chlorine (Cl2), phosgene, mustard gas, and hydrogen sulfide (H2S), making these cells a particularly important target for toxicity and for therapeutic intervention (Chauhan et al. 2008; Pita et al. 2013). A common feature of these gases is that acute exposure causes airway and lung epithelial cell apoptosis, alveolar-capillary barrier dysfunction, and an exacerbated inflammatory response (McGovern et al. 2011). Central to the pathophysiological mechanisms of these gases is the generation of ROS (Zhu et al. 2008). Many studies have proposed the use of antioxidants to diminish the effects of acute exposure to toxic gases as no effective antidotes or countermeasures have yet been discovered (Ji et al. 2010; Abel et al. 2011; Chauhan et al. 2008; Pita et al. 2013). Antioxidants like NAC and the metalloporphyrin AEOL10150 have been used with moderate success to reduce ROS, oxidative damage, and inflammation (McGovern et al. 2011; Ji et al. 2010). Few studies have investigated the beneficial effects of targeted Nrf2 activation to ameliorate toxic gas exposure, but this strategy may prove to be more effective.
Nrf2 also participates in infection-induced ALI. The bacteria Staphylococcus aureus causes pneumonia and in extreme cases progresses to ALI due to increased alveolar permeability, neutrophil infiltration and cytokine production. Nrf2−/− mice developed ALI after S. aureus inoculation, unlike Nrf2+/+ mice, due to enhanced inflammation and decreased mitochondrial biogenesis in alveolar cells of Nrf2−/− mice (Athale et al. 2012). Mitochondria not only maintain cellular energy production, but when damaged, they can also be a major source of ROS. Autophagy, in particular mitophagy, degrades damaged mitochondria. Therefore, these results suggest that the mitochondrial effects of Nrf2 and possibly its crosstalk with mitophagy are essential for lung adaptation and recovery from injury (Dinkova-Kostova, Abramov 2015).
The autophagy-lysosomal pathway
The autophagy-lysosomal pathway is a key cellular degradation pathway responsible not only for the removal of long-lived proteins, but also damaged or dysfunctional proteins and organelles, breaking them down to monomeric components for re-use by the cell. Autophagic dysfunction has been linked to the progression of a number of diseases including diabetes, neurodegeneration, cardiovascular disease, pulmonary disease, and cancer, demonstrating the importance of proper autophagic function in preserving normal cellular homeostasis (Choi et al. 2013). Three main autophagic pathways have been identified: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy (hereafter referred to as autophagy). The three main stages of the autophagy pathway are, (i) initiation, (ii) elongation, and (iii) fusion (Figure 1), and are mediated by a number of tightly regulated protein-protein and protein-lipid interactions. Initiation of autophagy requires formation of the Beclin-1/Vps34/UVRAG/Atg14L complex, which based on its binding partners, can result in either autophagy activation (i.e. bound to AMBRA-1 or Bif-1) or autophagy inhibition (i.e. bound to Rubicon or GAPR-1) (Kroemer et al. 2010; Levine et al. 2015). Following initiation, elongation of the phagophore involves the activation of two autophagy-related (Atg) protein-dependent ubiquitin-like conjugation systems, Atg5 conjugation to Atg12 mediated by Atg7 and Atg10, and the conjugation of microtubule-associated protein light chain 3 (LC3) to phosphatidylethanolamine via Atg3 and Atg10 (Ravikumar et al. 2010). The elongating phagophore encapsulates its cargo to form a double membrane-bound autophagosome, which then fuses with the lysosome via the interaction between endosomal sorting complexes required for transport (ESCRT) proteins, soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins, as well as Rab7 and a number of vacuolar protein sorting (Vps) proteins (Ravikumar et al. 2010). This fused vesicle, termed the autolysosome, can then degrade its contents using a battery of hydrolases, which break down the cargo for re-use by the cell.
Activation of autophagy during ALI
While the role of autophagy in chronic lung disease and cigarette smoke exposure has been studied (Chen et al. 2008; Ryter, Choi 2010), very few studies have investigated the role of autophagy in ALI. As mentioned above, ALI is induced by different stimuli, including mechanical injury, exposure to industrial or environmental toxicants, infection, and sepsis. Autophagy has been shown to contribute to the progression of acute viral and bacterial lung infection. Atg5fl/fl;LysM-Cre+ mice, which have autophagy deficient macrophages, exhibit increased bacterial burden and inflammation following infection with Mycobacterium tuberculosis (Castillo et al. 2012). Autophagy also plays a key role in alveolar macrophages and mast cells in response to Pseudomonas aeruginosa infection, with knockdown of Beclin1 or Atg5, two key autophagy initiation proteins, resulting in decreased clearance and intracellular accumulation of Pseudomonas in infected macrophages (Yuan et al. 2012). These studies indicate that autophagy can play either a protective or detrimental role, depending on the pathogen and cell type affected during lung infection.
One of the main initiators of autophagy in ALI is increased oxidative stress, which has been shown to induce autophagy in a number of lung cell lines and animal models of lung injury (Malaviya et al. 2014). For example, lung endothelial cells of rodents exposed to sulfur mustard or nitrogen mustard exhibit increased oxidative stress and autophagy activation within 6 h of gas exposure (Malaviya et al. 2010). O3 exposure has also been shown to result in oxidative stress-induced autophagy in the rat lung (Sunil et al. 2012). Heavy metal exposure, such as arsenic trioxide, can also lead to oxidative stress and autophagy dysregulation in human bronchial epithelial cells (Zhang et al. 2012). Exposure of H441 lung epithelial cells to chlorine gas (Cl2) induces mitochondrial dysfunction and increased reactive oxygen species production 1 hour after exposure, with increased autophagy being associated with improved bioenergetic function by 6 h post-exposure (Jurkuvenaite et al. 2015). While autophagy upregulation is thought to be sufficient for preventing toxic protein aggregation during mild oxidative stress, if the damage is too great and autophagy can no longer compensate, apoptosis is initiated leading to cell death (Chen et al. 2010). This may account for why autophagy upregulation is generally observed during lung injury, but is not always sufficient to mitigate or protect against inflammatory damage. Thus, a need for a better understanding of the temporal regulation of autophagy activation during ALI will be required to create therapeutics that maintain the beneficial effects of autophagy upregulation and reduce proteotoxic effects following toxic-gas exposure, and in turn, preserve lung function.
Protective role of autophagy during ALI
Studies have also demonstrated that upregulation of autophagy can be protective during ALI. For example, pharmacological activation of autophagy using rapamycin, a well-established autophagy activator, enhances the clearance of Pseudomonas from lung epithelial cells, whereas inhibition of autophagy using the lysosomal inhibitor chloroquine resulted in the accumulation and decreased killing of Pseudomonas bacteria (Junkins et al. 2013). Autophagy is downregulated following cecal ligation and puncture (CLP) injury, a model for sepsis-induced ALI, with rapamycin treatment enhancing autophagic activity, reducing the pro-inflammatory response, and preventing activation of the apoptotic cascade in septic mice (Yen et al. 2013). Transgenic mice overexpressing LC3, a protein integral for autophagosome formation, also exhibit improved survival following CLP (Lo et al. 2013). LPS-induced lung damage is exacerbated by the inhibition of autophagy, and rapamycin treatment decreased LPS-induced inflammatory cytokine production and secretion in lung macrophages (Lorne et al. 2009; Nakahira et al. 2011; Harris et al. 2011). Mice administered trehalose (2% in the drinking water), an mTOR-independent autophagy activator, for 6 weeks or aerosolized trehalose administration for 24 h prior to Cl2 exposure demonstrated improved bioenergetic function and decreased lung inflammation (Jurkuvenaite et al. 2015). Rapamycin treatment following paraquat-induced oxidative stress and ALI also significantly reduced lung inflammation and damage in mice via the inhibition of mTOR to activate autophagy (Chen et al. 2015). Interestingly, low dose carbon monoxide has also been shown to stimulate oxidative stress, which upregulates autophagy to protect against hyperoxia-induced lung injury (Lee et al. 2011). These studies indicate that autophagy activation, both before and after toxic insult, can significantly decrease lung inflammation, improve lung cell bioenergetic function, and mitigate the damage associated with ALI.
Dual activation of Nrf2 and autophagy as a potential therapy for ALI
As the above studies indicate, proper upregulation of Nrf2 and the autophagy pathway are required to mitigate damage during ALI, and thus represent two key pathways that can be targeted for therapeutic benefit. Since there is crosstalk between autophagy and Nrf2 activation through the Keap1-p62 interaction, it is presumable that dual pathway activators could be used to maximize the therapeutic benefit of Nrf2 and autophagy activation, however, the mechanism of activation must be considered.
In recent years, uncontrolled Nrf2 activation, the “dark side” of Nrf2, has been recognized as a contributing factor of cancer chemoresistance and tissue damage (Lau et al. 2008; Jiang et al. 2015). This uncontrolled activation may result in either constitutive or prolonged activation of Nrf2. The constitutive activation of Nrf2 observed in numerous cancer cell lines and patient tumor samples confers them a more malignant phenotype, as assessed by higher proliferative rates, increased migration, and chemo- or radio-resistance (Shibata et al. 2008; Tao et al. 2014; Hayes, McMahon 2009; Ooi et al. 2013; Ma et al. 2012; Hast et al. 2013; Wang et al. 2008; Chen et al. 2009). Prolonged Nrf2 activation occurs when autophagy is dysregulated, either by deletion of Atg5 and Atg7, or when autophagosome-lysosome fusion is blocked by arsenic treatment, causing the formation of p62-Keap1 aggregates and Nrf2 activation (Jiang et al. 2015). Autophagy dysfunction and prolonged Nrf2 activation cause tissue damage, inflammation, fibrosis, and tumorigenesis. In the lungs, inducible disruption of Atg7 caused bronchiolar epithelial cell edema and hyper-responsiveness of the lungs to a cholinergic stimulus (Inoue et al. 2011). Interestingly, these Atg7−/− cells had p62 accumulation and increased levels of Nrf2 target genes. On the other hand, basal or pharmacologically-induced autophagy, and the subsequent activation of Nrf2, may constitute a good approach for lung protection. This mode of autophagy activation, much like the canonical activation of Nrf2, is oscillatory: activation of autophagy occurs after a stimulus (for example, oxidative stress or rapamycin administration) and elimination of the stimulus (restoration of redox homeostasis or drug metabolism and excretion) restores the pathway to basal levels. Therefore, this controlled dual Nrf2-autophagy activation maintains the cytoprotective properties of both pathways.
Conclusion
ALI/ARDS are lung dysfunction syndromes with high prevalence and mortality, mainly because no therapies against them have been approved. ALI can be induced directly or indirectly by oxidative stress. Airway and lung epithelial cells are protected from oxidative stress by the Nrf2 and autophagy pathways. While extensive research has demonstrated the importance of Nrf2 in the protection against ALI and other lung pathologies, the role of autophagy is still ambiguous, though sufficient evidence supports its role as protective. Controlled dual activation of Nrf2 and autophagy may prove to be a better treatment strategy than single pathway activation due to the existing crosstalk of these pathways and potential synergistic beneficial effects.
Acknowledgments
This research was supported in part by the following National Institutes of Health grants: R01CA154377, R01ES015010, and R01ES023758 (DDZ); P01HL58064 (JGNG); HL60190, HL67841, and P01HL0101902 (SMB)
Footnotes
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
References
- Abel EL, Bubel JD, Simper MS, Powell L, McClellan SA, Andreeff M, et al. Protection against 2-chloroethyl ethyl sulfide (CEES)-induced cytotoxicity in human keratinocytes by an inducer of the glutathione detoxification pathway. Toxicol Appl Pharmacol. 2011;255(2):176–83. doi: 10.1016/j.taap.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Dimitropoulou C, Lu Q, Black SM, Sharma S. Glutathione supplementation attenuates lipopolysaccharide-induced mitochondrial dysfunction and apoptosis in a mouse model of acute lung injury. Front Physiol. 2012;3:161. doi: 10.3389/fphys.2012.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2(7511):319–23. doi: 10.1016/s0140-6736(67)90168-7. [DOI] [PubMed] [Google Scholar]
- Athale J, Ulrich A, Chou Macgarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, et al. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med. 2012;53(8):1584–94. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L, Lleres D, Swift S, Dinkova-Kostova AT. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc Natl Acad Sci U S A. 2013;110(38):15259–64. doi: 10.1073/pnas.1305687110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley MB. The pathologist’s approach to acute lung injury. Arch Pathol Lab Med. 2010;134(5):719–27. doi: 10.1043/1543-2165-134.5.719. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149(3 Pt 1):818–24. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- Bernard K, Hecker L, Luckhardt TR, Cheng G, Thannickal VJ. NADPH Oxidases in Lung Health and Disease. Antioxid Redox Sign. 2014;20(17):2838–53. doi: 10.1089/ars.2013.5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune B, Dehne N, Grossmann N, Jung M, Namgaladze D, Schmid T, et al. Redox control of inflammation in macrophages. Antioxid Redox Signal. 2013;19(6):595–637. doi: 10.1089/ars.2012.4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos R, Shimizu MH, Volpini RA, de Braganca AC, Andrade L, Lopes FD, et al. N-acetylcysteine prevents pulmonary edema and acute kidney injury in rats with sepsis submitted to mechanical ventilation. Am J Physiol Lung Cell Mol Physiol. 2012;302(7):L640–50. doi: 10.1152/ajplung.00097.2011. [DOI] [PubMed] [Google Scholar]
- Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109(46):E3168–76. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci U S A. 1999;96(22):12731–6. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Chauhan S, D’Cruz R, Faruqi S, Singh KK, Varma S, et al. Chemical warfare agents. Environ Toxicol Pharmacol. 2008;26(2):113–22. doi: 10.1016/j.etap.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Chen D, Ma T, Liu XW, Yang C, Liu Z. Rapamycin reverses paraquat-induced acute lung injury in a rat model through inhibition of NFkappaB activation. Int J Clin Exp Pathol. 2015;8(5):4627–38. [PMC free article] [PubMed] [Google Scholar]
- Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34(6):663–73. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10):e3316. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–5. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY. Genomic structure and variation of nuclear factor (erythroid-derived 2)-like 2. Oxid Med Cell Longev. 2013;2013:286524. doi: 10.1155/2013/286524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Gladwell W, Yamamoto M, Kleeberger SR. Exacerbated airway toxicity of environmental oxidant ozone in mice deficient in Nrf2. Oxid Med Cell Longev. 2013;2013:254069. doi: 10.1155/2013/254069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Gladwell W, Marzec J, McCaw ZR, Bienstock RJ, et al. Association of Nrf2 polymorphism haplotypes with acute lung injury phenotypes in inbred strains of mice. Antioxid Redox Signal. 2015a;22(4):325–38. doi: 10.1089/ars.2014.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, et al. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26(2):175–82. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- Cho HY, Kleeberger SR. Noblesse oblige: NRF2 functions in the airways. Am J Respir Cell Mol Biol. 2014;50(5):844–7. doi: 10.1165/rcmb.2014-0116PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Kleeberger SR. Association of Nrf2 with airway pathogenesis: lessons learned from genetic mouse models. Arch Toxicol. 2015 doi: 10.1007/s00204-015-1557-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Marzec J, Kleeberger SR. Functional polymorphisms in NRF2: implications for human disease. Free Radic Biol Med. 2015c doi: 10.1016/j.freeradbiomed.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8(1–2):76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32(32):3765–81. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordova EJ, Jimenez-Morales S, Centeno F, Martinez-Hernandez A, Martinez-Aguilar N, Del-Rio-Navarro BE, et al. NFE2L2 gene variants and susceptibility to childhood-onset asthma. Rev Invest Clin. 2011;63(4):407–11. [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015 doi: 10.1016/j.freeradbiomed.2015.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, et al. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy. 2010;6(5):614–21. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis RC, Vaporidi K, Bloch KD, Ichinose F, Zapol WM. Protective and Detrimental Effects of Sodium Sulfide and Hydrogen Sulfide in Murine Ventilator-induced Lung Injury. Anesthesiology. 2011;115(5):1012–21. doi: 10.1097/ALN.0b013e31823306cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–97. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hast BE, Goldfarb D, Mulvaney KM, Hast MA, Siesser PF, Yan F, et al. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 2013;73(7):2199–210. doi: 10.1158/0008-5472.CAN-12-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34(4):176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364(14):1293–304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- Howard MD, Greineder CF, Hood ED, Muzykantov VR. Endothelial targeting of liposomes encapsulating SOD/catalase mimetic EUK-134 alleviates acute pulmonary inflammation. J Control Release. 2014;177:34–41. doi: 10.1016/j.jconrel.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZS, Gu ZL, Sun MN, Zhang K, Gao PH, Yang QW, et al. Ursolic acid improves survival and attenuates lung injury in septic rats induced by cecal ligation and puncture. Journal of Surgical Research. 2015;194(2):528–36. doi: 10.1016/j.jss.2014.10.027. [DOI] [PubMed] [Google Scholar]
- Husari A, Khayat A, Bitar H, Hashem Y, Rizkallah A, Zaatari G, et al. Antioxidant activity of pomegranate juice reduces acute lung injury secondary to hyperoxia in an animal model. BMC Res Notes. 2014;7:664. doi: 10.1186/1756-0500-7-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51(5):618–31. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Inoue D, Kubo H, Taguchi K, Suzuki T, Komatsu M, Motohashi H, et al. Inducible disruption of autophagy in the lung causes airway hyper-responsiveness. Biochem Biophys Res Commun. 2011;405(1):13–8. doi: 10.1016/j.bbrc.2010.12.092. [DOI] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–91. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–91. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji L, Liu R, Zhang XD, Chen HL, Bai H, Wang X, et al. N-acetylcysteine attenuates phosgene-induced acute lung injury via up-regulation of Nrf2 expression. Inhal Toxicol. 2010;22(7):535–42. doi: 10.3109/08958370903525183. [DOI] [PubMed] [Google Scholar]
- Jiang T, Harder B, Vega MR, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med. 2015 doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junkins RD, Shen A, Rosen K, McCormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS One. 2013;8(8):e72263. doi: 10.1371/journal.pone.0072263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkuvenaite A, Benavides GA, Komarova S, Doran SF, Johnson M, Aggarwal S, et al. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med. 2015;85:83–94. doi: 10.1016/j.freeradbiomed.2015.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–23. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93. doi: 10.1016/j.molcel.2010.09.023. doi: S1097-2765(10)00751-3 [pii] 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008;58(5–6):262–70. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30(13):3275–85. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, et al. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol. 2013;33(12):2436–46. doi: 10.1128/MCB.01748-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim HP, Choi AM, et al. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol. 2011;45(4):867–73. doi: 10.1165/rcmb.2010-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Liu R, Dong X, Zhong Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015 doi: 10.1016/j.tcb.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingaraju MC, Pathak NN, Begum J, Balaganur V, Bhat RA, Ram M, et al. Betulinic acid negates oxidative lung injury in surgical sepsis model. J Surg Res. 2015;193(2):856–67. doi: 10.1016/j.jss.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Lo S, Yuan SS, Hsu C, Cheng YJ, Chang YF, Hsueh HW, et al. Lc3 over-expression improves survival and attenuates lung injury through increasing autophagosomal clearance in septic mice. Ann Surg. 2013;257(2):352–63. doi: 10.1097/SLA.0b013e318269d0e2. [DOI] [PubMed] [Google Scholar]
- Lorne E, Zhao X, Zmijewski JW, Liu G, Park YJ, Tsuruta Y, et al. Participation of mammalian target of rapamycin complex 1 in Toll-like receptor 2- and 4-induced neutrophil activation and acute lung injury. Am J Respir Cell Mol Biol. 2009;41(2):237–45. doi: 10.1165/rcmb.2008-0290OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol. 2012;32(8):1506–17. doi: 10.1128/MCB.06271-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Laskin JD, Laskin DL. Oxidative stress-induced autophagy: role in pulmonary toxicity. Toxicol Appl Pharmacol. 2014;275(2):145–51. doi: 10.1016/j.taap.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Sunil VR, Cervelli J, Anderson DR, Holmes WW, Conti ML, et al. Inflammatory effects of inhaled sulfur mustard in rat lung. Toxicol Appl Pharmacol. 2010;248(2):89–99. doi: 10.1016/j.taap.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamary AJ, Kondapaneni S, Vance GB, Gaughan JP, Martin UJ, Criner GJ. Survival in Patients Receiving Prolonged Ventilation: Factors that Influence Outcome. Clin Med Insights Circ Respir Pulm Med. 2011;5:17–26. doi: 10.4137/CCRPM.S6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzec JM, Christie JD, Reddy SP, Jedlicka AE, Vuong H, Lanken PN, et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007;21(9):2237–46. doi: 10.1096/fj.06-7759com. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. Journal of Clinical Investigation. 2012;122(8):2731–40. doi: 10.1172/Jci60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L379–99. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern T, Day BJ, White CW, Powell WS, Martin JG. AEOL10150: a novel therapeutic for rescue treatment after toxic gas lung injury. Free Radic Biol Med. 2011;50(5):602–8. doi: 10.1016/j.freeradbiomed.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20(7):1126–67. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, Kimura T, et al. Role of 15-deoxy delta(12,14) prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am J Respir Crit Care Med. 2005;171(11):1260–6. doi: 10.1164/rccm.200406-755OC. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi A, Dykema K, Ansari A, Petillo D, Snider J, Kahnoski R, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013;73(7):2044–51. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- Papaiahgari S, Yerrapureddy A, Reddy SR, Reddy NM, Dodd OJ, Crow MT, et al. Genetic and pharmacologic evidence links oxidative stress to ventilator-induced lung injury in mice. Am J Respir Crit Care Med. 2007;176(12):1222–35. doi: 10.1164/rccm.200701-060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pita R, Marco-Contelles J, Ramos E, Del Pino J, Romero A. Toxicity induced by chemical warfare agents: insights on the protective role of melatonin. Chem Biol Interact. 2013;206(2):134–42. doi: 10.1016/j.cbi.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–33. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol. 2012;32(17):3486–99. doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90(4):1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- Reddy NM, Kleeberger SR, Kensler TW, Yamamoto M, Hassoun PM, Reddy SP. Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J Immunol. 2009a;182(11):7264–71. doi: 10.4049/jimmunol.0804248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy NM, Potteti HR, Mariani TJ, Biswal S, Reddy SP. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am J Respir Cell Mol Biol. 2011;45(6):1161–8. doi: 10.1165/rcmb.2011-0144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy NM, Suryanaraya V, Yates MS, Kleeberger SR, Hassoun PM, Yamamoto M, et al. The triterpenoid CDDO-imidazolide confers potent protection against hyperoxic acute lung injury in mice. Am J Respir Crit Care Med. 2009b;180(9):867–74. doi: 10.1164/rccm.200905-0670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy NM, Suryanarayana V, Kalvakolanu DV, Yamamoto M, Kensler TW, Hassoun PM, et al. Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J Immunol. 2009c;183(7):4601–8. doi: 10.4049/jimmunol.0901754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol. 2010;191(3):537–52. doi: 10.1083/jcb.201005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Choi AM. Autophagy in the lung. Proc Am Thorac Soc. 2010;7(1):13–21. doi: 10.1513/pats.200909-101JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105(36):13568–73. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohrati M, Karimzadeh I, Saburi A, Khalili H, Ghanei M. The role of N-acetylcysteine in the management of acute and chronic pulmonary complications of sulfur mustard: a literature review. Inhalation Toxicology. 2014;26(9):507–23. doi: 10.3109/08958378.2014.920439. [DOI] [PubMed] [Google Scholar]
- Slutsky AS. Ventilator-induced lung injury: from barotrauma to biotrauma. Respir Care. 2005;50(5):646–59. [PubMed] [Google Scholar]
- Sun Z, Wu T, Zhao F, Lau A, Birch CM, Zhang DD. KPNA6 (Importin {alpha}7)-mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Mol Cell Biol. 2011;31(9):1800–11. doi: 10.1128/MCB.05036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Patel-Vayas K, Shen J, Laskin JD, Laskin DL. Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress. Toxicol Appl Pharmacol. 2012;263(2):195–202. doi: 10.1016/j.taap.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A. 2012;109(34):13561–6. doi: 10.1073/pnas.1121572109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Rojo de la Vega M, Quijada H, Wondrak GT, Wang T, Garcia JGN, et al. Bixin protects mice against ventilation-induced lung injury in an NRF2-dependent manner. Scientific Reports. 2015 doi: 10.1038/srep18760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74(24):7430–41. doi: 10.1158/0008-5472.CAN-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387(10–11):1311–20. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, et al. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol. 2007;27(21):7511–21. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve NF, Lau A, Zhang DD. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: an insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010;13(11):1699–712. doi: 10.1089/ars.2010.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkey AJ, Summer R, Ho V, Alkana P. Acute respiratory distress syndrome: epidemiology and management approaches. Clin Epidemiol. 2012;4:159–69. doi: 10.2147/CLEP.S28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, An J, Ji F, Jiao H, Sun H, Zhou D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun. 2008;373(1):151–4. doi: 10.1016/j.bbrc.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Warabi E, Takabe W, Minami T, Inoue K, Itoh K, Yamamoto M, et al. Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: role of reactive oxygen/nitrogen species. Free Radic Biol Med. 2007;42(2):260–9. doi: 10.1016/j.freeradbiomed.2006.10.043. [DOI] [PubMed] [Google Scholar]
- Ward HE, Nicholas TE. Alveolar type I and type II cells. Aust N Z J Med. 1984;14(5 Suppl 3):731–4. [PubMed] [Google Scholar]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–49. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Wilson MR, Takata M. Inflammatory mechanisms of ventilator-induced lung injury: a time to stop and think? Anaesthesia. 2013;68(2):175–8. doi: 10.1111/anae.12085. [DOI] [PubMed] [Google Scholar]
- Wright BJ. Lung-protective ventilation strategies and adjunctive treatments for the emergency medicine patient with acute respiratory failure. Emerg Med Clin North Am. 2014;32(4):871–87. doi: 10.1016/j.emc.2014.07.012. [DOI] [PubMed] [Google Scholar]
- Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014;28(7):708–22. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Sousse LE, Enkhbaatar P, Kraft ER, Deyo DJ, Wright CL, et al. gamma-tocopherol nebulization decreases oxidative stress, arginase activity, and collagen deposition after burn and smoke inhalation in the ovine model. Shock. 2012;38(6):671–6. doi: 10.1097/SHK.0b013e3182758759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S, Lowther S, Stambas J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J Virol. 2015;89(5):2672–83. doi: 10.1128/JVI.03529-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen YT, Yang HR, Lo HC, Hsieh YC, Tsai SC, Hong CW, et al. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery. 2013;153(5):689–98. doi: 10.1016/j.surg.2012.11.021. [DOI] [PubMed] [Google Scholar]
- Yilmaz MZ, Guzel A, Torun AC, Okuyucu A, Salis O, Karli R, et al. The therapeutic effects of anti-oxidant and anti-inflammatory drug quercetin on aspiration-induced lung injury in rats. J Mol Histol. 2014;45(2):195–203. doi: 10.1007/s10735-013-9542-3. [DOI] [PubMed] [Google Scholar]
- Yuan KF, Huang CH, Fox J, Laturnus D, Carlson E, Zhang BJ, et al. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. 2012;125(2):507–15. doi: 10.1242/jcs.094573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38(4):769–89. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23(22):8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24(24):10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Qi Y, Liao M, Xu M, Bower K, Frank J, et al. Autophagy is a cell self-protective mechanism against arsenic-induced cell transfomation. Toxicol Sci. 2012 doi: 10.1093/toxsci/kfs240. doi: kfs240[pii] 10.1093/toxsci/kfs240. [DOI] [PubMed] [Google Scholar]
- Zhao WC, Zhou SL, Yao WF, Gan XL, Su GJ, Yuan DD, et al. Propofol prevents lung injury after intestinal ischemia-reperfusion by inhibiting the interaction between mast cell activation and oxidative stress. Life Sciences. 2014;108(2):80–7. doi: 10.1016/j.lfs.2014.05.009. [DOI] [PubMed] [Google Scholar]
- Zhu L, Pi J, Wachi S, Andersen ME, Wu R, Chen Y. Identification of Nrf2-dependent airway epithelial adaptive response to proinflammatory oxidant-hypochlorous acid challenge by transcription profiling. Am J Physiol Lung Cell Mol Physiol. 2008;294(3):L469–77. doi: 10.1152/ajplung.00310.2007. [DOI] [PubMed] [Google Scholar]