

Abstract
The plasma kallikrein-kinin system (KKS) plays a critical role in human physiology. The KKS encompasses coagulation factor XII (FXII), the complex of prekallikrein (PK) and high molecular weight kininogen (HK). The conversion of plasma to kallikrein by the activated FXII and in response to numerous different stimuli leads to the generation of bradykinin (BK) and activated HK (HKa, an antiangiogenic peptide). BK is a proinflammatory peptide, a pain mediator and potent vasodilator, leading to robust accumulation of fluid in the interstitium. Systemic production of BK, HKa with the interplay between BK bound-BK receptors and the soluble form of HKa are key to angiogenesis and hemodynamics. KKS has been implicated in the pathogenesis of inflammation, hypertension, endotoxemia, and coagulopathy. In all these cases increased BK levels is the hallmark. In some cases, the persistent production of BK due to the deficiency of the blood protein C1-inhibitor, which controls FXII, is detrimental to the survival of the patients with hereditary angioedema (HAE). In others, the inability of angiotensin converting enzyme (ACE) to degrade BK leads to elevated BK levels and edema in patients on ACE inhibitors. Thus, the mechanisms that interfere with BK liberation or degradation would lead to blood pressure dysfunction. In contrast, anti-kallikrein treatment could have adverse effects in hemodynamic changes induced by vasoconstrictor agents. Genetic models of kallikrein deficiency are needed to evaluate the quantitative role of kallikrein and to validate whether strategies designed to activate or inhibit kallikrein may be important for regulating whole-body BK sensitivity.
Keywords: The plasma-kallikrein-kinin system (KKS), prolylcarboxypeptidase, prekallikrein, kallikrein inhibitors, high molecular weight kininogen, inflammation, sepsis
INTRODUCTION
The plasma kallikrein-kinin system (KKS) plays an important role in cardiovascular and cerebrovascular activities. The components of the KKS are part of a family of plasma proteins that function in vital overlapping physiological processes. Depending on the intensity of the stimulus, the activation of KKS leads to the activation of several sequential effector proenzymes resulting in the induction of genes and activation of biomolecules involved in the molecular mechanisms of vasodilation, blood coagulation, and fibrinolysis. Although the KKS function was initially recognized during tissue injury, little is understood about how the KKS activation occurs on vascular endothelial cells. The progressive elucidation of the presence and distribution of the KKS and its effectors within the cardiovasculature has provided the exact hepatocyte-cardiovascular correlates related to abnormal and normal cardiovascular physiology. The clearly defined roles of the KKS are to stimulate endothelial nitric oxide generation and to potentiate the activation of thrombin-induced clot formation.
Whereas the normally functioning KKS can successfully counteract blood pressure, promote smooth blood flow, modulates neovascularisation and eliminates pathogens by promoting the recruitment of neutrophils to the site of injury, inappropriate responses can lead to extensive tissue damage (hyperactivity). The focus of this review is to describe the cascade of molecular events that have been uncovered in both basic and clinical studies, which are directly relevant to understanding the KKS function in cardiovascular systems and may provide insights into mechanisms that directly contribute to the pathogenesis of tissue injury, organ failure, and death. Further knowledge about the interrelation of the KKS molecules, their metabolites which impart function to the cell-specific receptors, regulatory mechanisms, and cell-specific expression of genes may yield new approaches to improve patient outcome after vascular damage or surgical intervention.
SYNTHESIS AND STUCTURE OF THE KKS
The KKS consists of two functionally distinct complexes, specified by one molecule of prekallikrein (PK) in complex with one molecule of high molecular weight kininogen (HK) [HK/PK], or one molecule of factor XI (FXI) in complex with two molecules of HK [HK/FXI][1–4]. The physiological roles of HK and PK are being discussed here. For additional extensive and excellent reviews on the KKS, please read the following references[2,5–8].
High molecular weight kininogen synthesis, distribution and its biological functions
Chromosomal localization of the human plasma kininogen gene has been mapped to the 3q26 – qter region[9]. A single kininogen gene consisting of 11 exons transcribes a unique mRNA for high molecular weight kininogen (HK) and low molecular weight kininogen (LK) by alternative splicing[2]. HK is a single-chain plasma glycoprotein (110–120 kDa) primarily expressed in the liver and secreted into the bloodstream.
HK is a multifunctional protein composed of six domains with each domain believed to have distinct functions [2]. The heavy chain (64 kDa) of HK contains domains 1, 2, and 3 while the light chain (45 to 58 kDa) is comprised of domains 5 and 6[10]. The heavy chain and light chain are flanked by domain 4, which is ultimately liberated as BK. Domain 1 has a low affinity calcium-binding site, whereas domains 2 and 3 have specific sequences (Gln-Val-Val-Ala-Gly) that serve as inhibitors of tissue cysteine proteases[2,11]. Domain 3, like domain 5, has platelet- neutrophil- and endothelial cell-binding action [12,12–15]. Intact HK, but not the oxidized HK or its domain 3, is an inhibitor of cathepsin B and H[16,17].
It has been demonstrated that HK could bind to anionic charged surfaces through the histidine-glycine-lysine rich regions of the light chain corresponding to domain 5[18]. The binding of HK to factor XI or prekallikrein is mediated by a sequence comprising 31 amino acid residues in domain 6[3,19]. The ability to bind to a surface and simultaneously complex PK and factor XI results from the cofactor activity of HK in the contact activation pathway [20,21].
Early interests in a variety of diseases led researchers to actively pursue basic studies into the assembly of HK/PK in the central nervous system and many other tissues. Das and others[22] demonstrate the colocalization of HK with amyloid precursor protein (APP) near cerebral blood vessels, in the neutrophil, and in many neurons of the rat brain, suggesting that the interaction of HK with APP might modulate neurogenesis.
The BK-free-HK [cleaved HK (HKa)] is an anti-angiogenic agent, implicating its ability to modulate BK –induced angiogenesis signaling[23]. The role of HKa in endothelial cell migration, proliferation and tube formation is accepted; however, any physiological benefit on microvascular structure requires further investigations. Future clinical trials to support the efficacy of HKa or anti-HK antibody as a therapeutic regimen to inhibit angiogenesis and tumor growth in human therapy are anticipated.
Kallikrein generation, distribution and its biological actions
Chromosomal localization of the human plasma kallikrein gene has been mapped to the q34 – q35 region on the long arm of chromosome 4, encoded by a single gene[24]. It is synthesized predominantly in the liver as a proenzyme, prekallikrein, also known as prokallikrein. Human plasma prekallikrein is synthesized as a precursor with a signal peptide of 19 amino acids. The mature form of the protein which circulates in the blood is a single-chain polypeptide of 619 amino acids[25,26]. Activation of prekallikrein to kallikrein occurs through a cleavage at the Arg371-Ile372 producing a two-subunit protein involving a heavy chain and a light chain, linked through a disulfide bond between Cys364 and Cys484. The N-terminal heavy chain of 371 amino acids contains four apple domains whose homologues are also found in factor XI (FXI); whereas, the C-terminal light chain forms the catalytic domain[27]. Plasma prekallikrein associates with high molecular weight kininogen (HK), an α-globulin which circulates in plasma as an 88- to 120-kDa single-chain glycoprotein at a concentration of 70 to 90 μg/mL[28].
Functional plasma kallikrein (activated PK) can be generated on blood vessel endothelial cells as a result of the HK/PK activation[29]. Plasma kallikrein is implicated in many physiological and pathological processes including blood coagulation, the initiation of the classical complement cascade pathway, as well as activation of the alternative complement pathway[30–33]. Plasma kallikrein is also involved in induction of elastase release from neutrophils and conversion of prourokinase to urokinase to initiate fibrinolysis[34–37]. Plasma kallikrein hyperactivation parallels endothelial lesion, tissue injury, and sepsis, underscoring the correlation between plasma kallikrein alterations and inflammation [38–40].
In-vivo and in-situ cellular imaging characterization of novel plasma kallikrein activity on endothelial and inflammatory cells, foamy macrophages and fibroblasts within the thickened intima of the plaque as well as in smooth muscle cells suggest that plasma kallikrein activity may be induced by atheromatous disease.[41]. In the study conducted using a patchy atheromatous disease, plasma kallikrein and PK are present in the coronary, vertebral and supracallosal arteries, suggesting that atheromatous disease triggers the activation of PK. Immunolabelling of the components of the plasma KKS in blood vessels shows the presence of circulating prekallikrein/plasma kallikrein in plasma as well as on the endothelial cells of the medium to small size arteries[41]. Interestingly, no trace of immunoreactive PK is detected in the renal vein at all[41]. This is significant because it highlights the physiological role of plasma kallikrein in particular tissues in animals.
The plasma KKS participates in the surface-dependent activation of blood coagulation
The coagulation cascade has long been thought of as a “water fall” of activating proteases initiated by the extrinsic and the intrinsic paths, which lead to the common pathway and ultimately the activation of thrombin and fibrin formation [Fig. (1)] [42]. The starting point for the extrinsic path is the complex between factor VIIa and tissue factor initiated through vessel damage. This starting point enters directly into the common pathway by activating factor X and to some extent factor IX, both of which lead to the activation of thrombin and ultimately clot formation. The classical view of the intrinsic pathway has been thought of as secondary since it is factor XII that activates Factor XI. Next, factor XI leads to the activation of factor IX which subsequently leads into the common pathway by the activation of Factor X and then thrombin [Fig. (1)] [42,43]. Now, with the recent identification of activities of the coagulation components, mainly around the contact pathway, the clotting cascade is being examined in alternative ways [Fig. (2)] [44]. One way to consider it is that both the contact pathway and the extrinsic pathway are starting points of the intrinsic pathway, which is now viewed as the system that propagates and stabilizes the newly initiated clot. Looking at it this way you can see that during acute vessel damage the extrinsic pathway has the potential to provide a relatively short burst of thrombin, 0.5 to 1nM[45]. This burst in thrombin may lead to the initiation of the “intrinsic amplification loop” (IAL) leading to increased activation of platelets and factor XI, resulting in the sustained production of thrombin and its contributors to coagulation[18]. While the contact system can be viewed in this way, the initiation of the IAL may take place over long periods of time such as in venous thrombosis. It can be hypothesized that these two initiating systems would be activated simultaneously during certain disease states, as in the event of plaque rupture in atherosclerosis. It is in such disease states that a robust burst of circulating inflammation markers appear to play major roles in the outcome of such events. This is where the role of the contact pathway and its proteases are believed to play major contributing roles, not only in thrombosis but in the inflammatory response as well.
Figure 1.
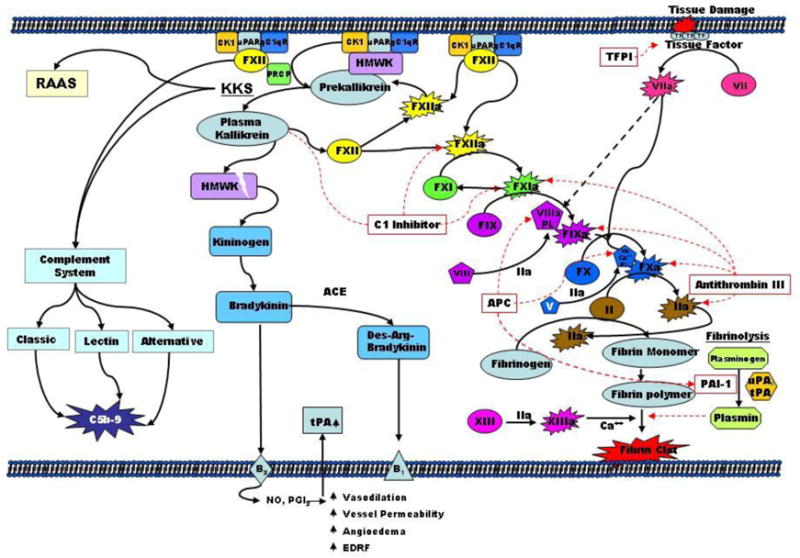
classical coagulation cascade. The blood coagulation cascade consists of an extrinsic and intrinsic pathway. Both pathways results in activation of factor X, which subsequently converts prothrombin to thrombin. The coagulation cascade is a starting point for both the kallikrein-kinin system (KKS) and the complement system. Activation is indicated by solid arrows and inhibition by dotted arrows.
Figure 2.
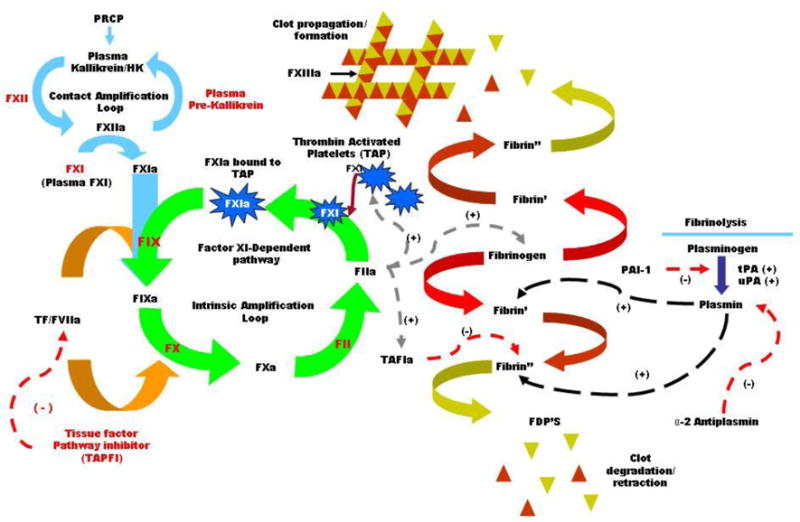
Amplification loops. The amplification loops associated with the coagulation cascade. The contact and intrinsic pathways make up the main amplification loops in the coagulation cascade. Activation is indicated by (+) and inhibition by (−).
Plasma kallikrein is a serine protease which plays a central role in the contact activation and kinin generation pathways[46]. For years the conjecture has been that the contact pathway is fairly benign in its physiological role in coagulation, in part because it was believed that it took an artificial negatively charged surface to activate it. However, new investigations into the role of the contact pathway have led to the discovery that the contact pathway is actually involved in many mechanisms involving inflammation, with the liberation of BK from HK, to the activation of the complement system and in the fibrinolytic pathway of hemostasis[47]. Once plasma kallikrein is activated from the zymogen precursor prekallikrein (Fletcher Factor), it initiates surface-mediated activation of coagulation, fibrinolysis and kinin generation [Fig. (1)] [47]. The activated plasma kallikrein is controlled primarily by endogenous C1 inhibitor as well as α2-macroglobulin and antithrombin III [Fig. (1)] [48]. An imbalance between plasma kallikrein and its naturally occurring plasma inhibitors is associated with several disease states, such as hereditary angioedema (HAE), inflammatory bowel disease (IBD), systemic lupus, rheumatoid arthritis (RA) and allergic rhinitis among others[11,49].
KKS activation and its effects on the cardiovascular system
The activation of the HK/PK triggers the release of the vasoactive peptide bradykinin (BK) and anti-angiogenic cleaved HK (HKa) from the high molecular weight kininogen by plasma kallikrein [50–55]. The mechanisms of assembly and activation of KKS in the cardiovascular system is still in its infancy. Recent postulations in the physiological regulation of KKS are here being described.
The autoactivation of FXII to activated factor XII (FXIIa) occurs after FXII bound to a negatively charged surface[56]. Of note, the rate of conversion of FXII to FXIIa is about 100 times faster in the presence of PK and HK, suggesting the presence of reciprocal proteolytic activation of FXII and PK[57]. Cochrane and Griffin[57] provide evidence that FXIIa is present in two forms, namely αFXIIa and βFXIIa. αFXIIa activates PK on the surface while βFXIIa cleaves PK in plasma (fluid phase). Furthermore, both α - and βFXIIa can activate PK while αFXIIa is a much better activator of FXI than βFXIIa [57].
Recent evidence suggests that the hyaluronan-binding protease (PHBSP, a plasma enzyme) known to activate factor VII (FVII) and pro-urokinase could also interact with HK/PK[58]. Surprisingly, these studies indicated that PHBSP activates PK to plasma kallikrein. The activation of PK causes the cleavage of HK and liberation of BK in the absence of any charged surface, which suggests a cytoprotective role of PHBSP-induced PK activation[58]. The authors suggest that the assembly of HK/PK complex on a negatively charged surface is not a prerequisite for PHBSP to liberate BK from HK, however, the physiologic and pathophysiologic significance of PHBSP-stimulatory pathway has not been pursued.
Joseph and Kaplan observed that heat shock protein 90 (HSP90) activates PK to plasma kallikrein, implicating a novel function for HSP90[59]. We find this observation interesting because it seems that a physiological change like stress would have a special role as an indicator of PK activation. However, the true nature of PK (a plasma protein) activation by HPS90 (a cytosolic/membrane protein) requires further investigations to determine their transmembrane movements that are restricted primarily by a phospholipid bilayer. The above observations characterize the complexity of possible interactions between HSP90 and the KKS.
Finally, PK associates with HK to a multiprotein receptor complex comprising in at least cytokeratin 1, gC1qR, and urokinase plasminogen activator receptor (u-PAR). It is this complex which enables the protease prolylcarboxypeptidase (PRCP) to activate PK. This model of PK activation is referred to as the factor XII independent pathway and can occur on extracellular matrix and dysfunctional endothelial cells [Fig. (1)][60–62]. The activation of PK by factor XIIa occurs on both membrane surfaces, where HK and PK are complexed together, and the plasma fluid phase where activation of PK can occur by a fragment of factor XII (βFXIIa)[63,64]. Once PK is activated to plasma kallikrein, it can then activate factor XII to its active form completing the contact amplification loop [Fig. (1)][65]. In vivo activity of plasma kallikrein has been shown to be relativity short lived due to its main inhibitor the serpin C1 inhibitor (C1-INH) [Fig. (1)][66].
In summary, these studies strongly demonstrate that HK/PK can be activated by a modest array of molecules in plasma, luminal cells, extracellular matrix and dysfunctional and functional endothelial cells. This may indicate that HK/PK is an alternate mechanism for a host to detect the onset of abnormalities within the blood vessel to initiate and coordinate wound healing processes. This is consistent with in-vivo findings, where the activation of HK/PK has been shown to promote wound healing.
KKS FUNCTION
The proteins of plasma HK/PK are unique in that they exert effects in most tissues of the cardiovascular system throughout the life of an individual. Many actions of the HK/PK are mediated through the effects of plasma kallikrein, HKa and BK to stimulate production of intra- and extracellular messengers and effectors in a variety of cells[67,68]. There is a large body of evidence providing proof that plasma KKS drives regional blood flow via BK-induced B2 receptor activation and promotes intravascular thrombus formation via factor XI activation after an injury.
The plasma KKS activation increases endothelium-dependent vasodilation and local blood flow
The plasma KKS is a vasodilator and contributes to the modulation of vasodepressor components of the cardiovascular system[69]. Plasma kallikrein’s association with the contact pathway provides one of several ways that KKS is involved directly or indirectly in thrombosis [Fig. (1)]. In looking at the primary role of plasma kallikrein in the liberation of BK from HK, the theory is that cleavage of HK (Mr=120, 000) by plasma kallikrein is a three-step reaction[70,71]. When HK is hydrolyzed by plasma kallikrein, cleavage first occurs at the C-terminus Arg-Ser end of the BK sequence of HK. This initial nick cleavage produces a stable intermediate followed by the cleavage of the N-terminus end of the BK sequence leading to the liberation of BK [Fig. (2)][72]. The result is two active peptides that play a role in both the inflammatory and coagulation process at the vasculature level.
Clarification of the precise mechanisms involved in the fine tuning of BK liberation, signaling, and degradation has contributed to understanding the etiology of inflammation and other related inflammatory disease states and aided in clinical treatment of some of these endocrinopathies [73–75]. Bradykinin participates in the etiology of vascular inflammation and the regulation of the microvascular circulation. Bradykinin stimulates endothelial cells to release a number of powerful mediators via the bradykinin B2 receptor (BKB2R) and bradykinin B1 receptor (BKB1R) activation [Fig. (2)]. Bradykinin is not only a critical player in promoting angiogenesis by stimulation of VEGF formation through BKB2R, but also plays a role in causing the upregulation of bFGF via BKB1R activation, thus strongly suggesting a genomic level of action of BK in neovascularization[76].
Angiotensin converting enzyme further digests full length BK into des-Arg BK which interacts with BKB1R [Fig. (2)][77]. The physiological/pathophysiological effect of BK on hemostasis has been investigated by several groups. In recent studies using BKB2R knock out mice it was demonstrated that they exhibited an increase time to occlusion in a mild thrombosis injury model but not in a more server thrombosis model[78]. It was postulated in this study that the interactions between the AT2R and the B2 receptor may interact and lead to an increase in expression of AT2R. An increase in AT2R may lead angiotensin to favor this receptor over AT1R and cause an elevation of NO and prostacyclin levels[78]. Studies looking at rat models that express both BK receptors show, in vitro, that BK acting through the B2 receptor on the surface of endothelial cells promotes the expression of procoagulant and antifybrinolytic proteins, such as tissue factor (TF) and plasminogen activator inhibitor 1 (PAI-1)[79]. This effect translated well to the human model described in the study using a selective bradykinin B2 receptor antagonist, which demonstrated that inhibition of BK activity on the B2 receptor resulted in a decrease in vasodilatation and t-PA release, which may cause a decrease in fibrinolytic activity[79–81].
The plasma KKS keeps the blood flowing
There are two distinct pathways that initiate the fibrinolysis pathway. In the first, described above, BK is released from HK and ultimately results in increased expression of PAI-1 and TF [Fig. (2)]. The second avenue of activating fibrinolysis involves the activity of plasma kallikrein on the conversion of pro-urokinase to urokinase. Early investigations found that urokinase plasminogen activator (u-PA) bound to the cell surface was able to activate plasminogen to plasmin as well as to auto-activate itself, generating yet another possible amplification loop[82]. It is postulated that the binding of HK and subsequently PK aligns pro-urokinase in such close proximity as to drive its activation in spite of the presence of PAI-1[Fig. (2)][83]. These studies indicate there is a pathway that activates plasminogen independent of tissue plasminogen activator (t-PA) and that plasma kallikrein plays a key role in the fibrinolysis system.
An additional way the KKS can effect thrombosis is the interactions of HKa and the effect this has on adhesion molecules. When plasma kallikrein liberates BK from HK, the remaining product is two-chain high molecular weight kininogen (HKa). HKa has been shown to compete with fibrinogen and adhesion molecules in binding to cell membranes through the “Vroman effect”, by which higher mobility proteins are replaced by lower mobility proteins with a greater affinity for the cell surface[84,85]. The process in which HKa could displace fibrinogen may be another way for plasma kallikrein, indirectly, to effect the fibrinolysis process. Investigations into the rare genetic trait, Fletcher Factor deficiency, demonstrated that these individuals exhibit abnormalities in coagulation, fibrinolysis and BK production with chemotacis abnormalities as well [Table 1][86]. In 1965 hereditary plasma prekallikrein deficiency was diagnosed in a 71-year-old man, for whom Fletcher Factor was named, by Hathaway, Belhasen and Hathaway [Table 1][87]. Evidence exists which has led to the speculation that Mr. Fletcher died of a possible thrombotic event. Subsequently, several mutations in the amino acid make up of plasma kallikrein have been identified however, these deficiencies and mutations have not been associated with any clinically severe thrombotic events[88,89]. There are also case reports which suggest that Fletcher Factor deficiency may be a risk factor for myocardial infarctions, even though biomarkers such as activated partial thromboplastin time is prolonged, indicating an antithrombotic effect [Table 1][90,91]. In both of these examples, other predisposing conditions were either not reported or not taken into consideration, making it difficult to ascertain weather or not inhibition or deficiencies of plasma kallikrein is beneficial or detrimental.
Table 1.
Characteristics of components of the plasma kallikrein-kinin system in cardiovascular diseases
| KKS components | Genetic manipulation | Species | Clinical Presentation | Demography | Peak blood level
|
Activity (%) | Ref | |||
|---|---|---|---|---|---|---|---|---|---|---|
| HK | HKa | K | FXIIa | |||||||
| HK | KO | Mouse | ↑aPTT | |||||||
| KO | Rat | ↑aPTT | – | 0.00 | – | N | – | 0.00 | [178] | |
| Deficiency | Rat | – | – | ↓ | – | – | – | ↓ | [179] | |
| Deficiency | Rat | ↑aortic aneurysm | ||||||||
| Deficiency | Rat | Risk factor for ischemic heart, impaired LV function | – | ↓ | – | – | – | – | [180] | |
| – | ↓ | – | – | – | – | [181] | ||||
| Deficiency | Rat | Reduced PK | ↓ | – | – | – | – | [182] | ||
| ↑Blood pressure, protected from inflammation, ↑aPTT | – | ↓ | – | – | – | – | [183–185] | |||
| KO | Hamster | Muscular dystrophy | – | ↓ | – | – | – | 69 | [186] | |
| KO | Human | ↑aPTT | female | <1% | ↓ | – | – | – | [187,188] | |
| Mutation | Human | NT | NK | ↓ | – | – | – | 0.00 | [189] | |
| KO | Human | VT | Australian | 0.00 | – | – | – | – | [190] | |
| Deficiency | Human | NT, ↓Vascular permeability | Caucasian | ↓ | – | – | – | ↓ | [191,192] | |
| Deficiency | Human | NT | Japanese | ↓ | – | – | – | – | [193,194] | |
| KO | Human | ↓Blood Coagulation | Japanese | ↓ | – | – | – | – | [195] | |
| KO | Human | ↑aPTT, depressed intrinsic fibrinolytic activity, ↑aPTT | Japanese | ↓ | – | – | – | – | [88] | |
| KO | Human | – | ↓ | – | – | – | – | [196] | ||
| Portuguese | ↓ | – | ↓ | N | – | [197] | ||||
| KO | Human | ↓BK receptor | – | ↓ | – | – | – | – | [198] | |
| KO | Human | Abnormal aPTT | Female twins | ↓ | – | – | – | – | [199] | |
| Prekallikrein | ||||||||||
| KO | Human | ↑aPTT, osteomyelofibrosis | Male | – | – | <2% | 5 | [200,201] | ||
| Deficiency | Human | – | – | 0.00 | – | ↓ | – | – | [202] | |
| Deficiency | Human | VT | Caucasian | – | – | ↓ | – | – | [191] | |
| Heterozygous | Human | ↓PK, ↑aPTT | Caucasian | – | – | ↓ | – | – | [203] | |
| KO | Human | ↓BK | – | – | – | ↓ | – | – | [204] | |
| KO | Human | Male | – | – | ↓ | – | – | [205] | ||
| Factor XI | ||||||||||
| Deficiency | Human | Caucasian | – | – | – | – | – | [201] | ||
| Homozygous | Human | ↑aPTT | – | – | – | – | – | – | [206] | |
| KO | Human | ↑aPTT | – | – | – | – | – | – | [207] | |
| Deficiency | Human | hemorrhage | Ashkenazi | |||||||
| Bleeding diathesis | Jews | – | – | – | – | – | [208] | |||
| Factor XII | ||||||||||
| KO | Mouse | ↓BK | – | N | N | N | ↓ | [209] | ||
| KO | Human | Thrombosis | Male | – | – | – | – | – | [39] | |
| Deficiency | Human | No phenotype | Female/Male | N | N | ↓ | ↓ | – | [210] | |
| Deficiency | Human | Myocardial infarction | – | – | – | – | ↓ | – | [211] | |
| Deficiency | Human | ↓PK activity | – | – | – | ↓ | – | – | [212] | |
| C1-inhibitor | ||||||||||
| KO | Human | HAE | – | ↓ | ↑ | ↑ | ↑ | – | [213–215] | |
| KO | Human | HAE | Pregnant | – | ↑ | ↑ | ↑ | – | [216,217] | |
| KO | Human | ↑Thrombin & BK generation |
– | – | – | – | – | – | [214] | |
HK: High molecular weight kininogen, HKa: Cleaved high molecular weight kininogen, VT: venous thrombosis, K: kallikrein, HAE: Hereditary angioedema, BK: Bradykinin, KO: knockout, NT: not tested, NK: not known, N: normal
The majority of evidence for the KKS role in hemostasis points to antithrombotic as well as prothrombotic, depending on the underlying cause of the event. One can imagine that during disease states which require the dissolution of clots through the fibrinolytic pathway, plasma kallikrein and the rest of the contact pathway would be a benefit. This paradox may be explained in part because of plasma kallikrein’s role in the fibrinolytic pathway, where plasma kallikrein may play a greater role, than in the coagulation pathway. This is not to say that the KKS and the contact system do not play pivotal rolls, quite the contrary. It is believed that these systems play a major role in hemostasis which involves the inflammatory system. The KKS contributes to various cardiovascular diseases which will be described in the following sections. Most of these articles point out that the KKS and contact system play roles in both inflammatory and thrombosis conditions [Table 1][8,62,92].
Therapeutic potential of kallikrein inhibitors in cardiovascular diseases
The KKS pathway is crucial not only in physiological but also in pathophysiological and progression of various cardiovascular diseases such as inflammation and hypertension, both of which are related to derangement of the vascular endothelial cells. The following sections summarize the evidence of in vitro, in vivo and clinical studies whereby plasma kallikrein and its potential inhibitors represent possible new approaches for the management of vascular diseases.
ROLE OF THE PLASMA KKS IN CARDIOVASCULAR ABNORMALITIES
Sepsis
Hypotensive bacteremia is associated with formation of plasma kallikrein and rise of plasma BK levels[93]. In sepsis, the activation of KKS results in increased plasma kinin levels and increased circulating kallikrein-C1 inhibitor complexes [94–96]. Sepsis is a multifactorial condition which promotes the release of BK as well as molecules that stimulate coagulation which left uncontrolled can lead to organ failure and death.
Sepsis is a progressive disease, usually presenting as a bacterial infection, and accounts for one in five of all hospital admissions. The term systemic inflammatory response syndrome (SIRS) is used to encompass conditions of sepsis that are not brought on by infection. For example, trauma burns and pancreatitis[97]. There are approximately 500,000+ cases of severe sepsis a year[98]. Risk factors contributing to the development of sepsis are: age, diabetes, cancer, HIV and surgical procedures. In general, any condition that increases the risk of infection or the need for surgical procedure can put an individual at risk for sepsis[99,100]. Studies have shown that the inflammatory system becomes active resulting in the appearance of both cellular components of the innate immune response and cytokines like TNF, IL-6, IL-1 and IL-8. Sepsis is likewise responsible for the shifting of the haemostatic state of coagulation towards a prothrombotic state, often resulting in disseminated intravascular coagulopathy (DIC)[101]. It is well documented that one of the responses to septic shock is the increase in vasodilatation resulting in hypotension, despite fluid resuscitation. This, combined with increased activation of both the inflammation and prothrombotic pathways, leads to severe tissue damage and multi-organ failure[102–104]. The activation of the KKS and subsequently the liberation of BK contributes significantly to the hypotensive effects of sepsis, partially by the secondary release of other mediators like nitric oxide and platelet activating factor[105]. Additional evidence for the participation of the KKS are reports that suggest the inducible BK receptor, B1, are up regulated during inflammatory and immunipathology states. This up regulation of B1, along with the actions of the constituently expressed B2 receptors, most likely contribute to the hypotensive state seen during septic shock. Indeed, studies using B2 antagoniost have shown partial therapeutic effects. However, it remains to be determined if a greater effect may be seen using a B2 and B1 antagonist or a nonselective antagonist[106].
Treatments for Sepsis
The primary focus on the treatment of sepsis is identification of the source with immediate administration of the proper antibiotics. Further treatments involve fluid resuscitation, vasopressors, inotropic therapy, steroids and recently recombinant human activated protein C therapy[107]. Recently, a great deal of research has been put into finding innovative and improved treatments for sepsis. Some of these have had positive results while others have had negative results associated with them [Table 2][98,102]. The pro-inflammatory pathway, including TNFα, interleukin 1 and BK, has been looked at with no real positive results[98]. However, in the case of BK antagonist treatment, investigators suggest there may be a benefit with this antagonist but further studies are warranted[108]. An earlier study, using a rat sepsis model treated with a BK and leukocyte inhibitor, demonstrated a possible synergistic survival benefit to sepsis. However, when a BK inhibitor alone was administered the results showed very little benefit compared to control groups[109]. This suggests that a plasma kallikrein inhibitor may be a possible target given the effect of plasma kallikrein on both BK liberation and on leukocyte adhesion. Corticosteroid treatment of severe sepsis has been one of the most perplexing approaches in the treatment of sepsis. Persuasive proof that corticosteroids are useful pharmacologic agents in the treatment of sepsis remains elusive despite numerous studies over the past four decades[94]. Recently, evidence suggests that treating sepsis with high doses of corticosteroids for relatively short periods of time is ineffective. However, there have been some encouraging results from recent trials using low doses of corticosteroids over longer periods of time, but these finding are contingent on meeting other criteria such as adrenocorticotropin hormone tests and crotisol concentrations[110]. Lately, recombinant human activated protein-C (drotrecogin alfa) has been evaluated in patients presenting with sepsis[111]. Activated protein C (APC) is an endogenous inhibitor of Va, VIIIa, and plasminogen activator inhibitor-1 (PAI-1) thereby functioning as an anticoagulant and profibrinolytic agent. APC has also been shown to exert anti-inflammatory effects by inhibiting cytokine production in monocytes (TNF-α, IL-1 and IL-6) and reducing adhesive interactions between neutrophils and endothelial cells[112,113]. Even though this is an agent which has been approved in the US by the food and drug administration, it has a limited therapeutic index. Strict adherence to exclusion criteria, as put forward by the PROWESS study, should be used in order to decrease the number of severe bleeding events[114]. Certain nonselective inhibitors of serine proteases with inhibitory activity in the inflammatory and coagulation pathways has been shown to exhibit some benefit in sepsis. One example of this, aprotinin, given during septic shock, was able to preserve vascular resistance and maintain arterial pressure while at the same time decreasing pulmonary pressure and renal function during sepsis. Despite the affects of aprotinin on the vasculature during septic shock, aprotinin has shown some adverse effects including inadequate coagulation and allergic reactions[115,116]. Due to the relatively low therapeutic index of recombinant human activated protein-C and the risk of adverse events with the use of aprotinin, additional treatments targeting both the inflammatory and coagulation systems should be investigated. For this reason, plasma kallikrein is a potential target for the treatment of sepsis and may add some benefit, as activated PK is the primary cause for the liberation of BK and has been shown to be an activator of the contact system.
Table 2.
Kallikrein inhibitors with structural diversity and varying potency reported in the literature
| Organization | Drug Name | Structure | Highest Phase | MOA | Condition | Basic Patent |
|---|---|---|---|---|---|---|
| Fovea,Cubist: Dyax | DX-88 |

|
Recommended Approal | Plasma Kallikrfein inhibitor | Surgery, arterial coronary; Angioedema, hereditary; Edema, macular | US 2006183771; WO 2006036860 |
| Showa Denko | PKSI-527 |
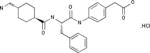
|
Preclinical | Plasma Kallikrein inhibitor | Thrombosis | EP 0217286 |
| Daiichi Sankyo |
|
Biological Testing | Plasma Kallikrein inhibitor | Pancreatitis | JP 1992159261 | |
| Novo Nordisk |

|
Biological Testing | Plasma Kallikrein inhibitor | Pancreatitis | US 5373090 | |
| Bristol-Meyers Squibb |
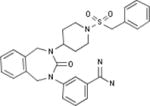
|
Biological Testing | Kallikrein 1 (Plasma Kallikrein) Inhibitors; Inhibitors of Blood Coagulation Pathways | Thrombosis | WO 1997038984 | |
| Celera |
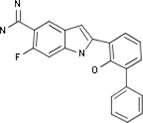
|
Biological Testing | Plasmin Inhibitors; Urokinase (u-PA) Inhibitors; Kallikrein 1 (Plasma Kallikrein) Inhibitors | Thrombosis | WO 2000035886 |
Source: Prous Integrity
Inflammation
The activation of the KKS results in the release of vasoactive bradykinin (BK), which modulates vascular biology[117]. BK exerts various biological and physiological responses such as vascular permeability elevation, smooth muscle contraction in the arteriole segment of the blood circulatory system, modulation of kidney function, pain sensation and induction of hypotension. BK causes vasodilatation by the stimulation of endothelial B2 receptors of arteries and arterioles, with subsequent endothelial release of nitric oxide and prostaglandins[117]. BK influences the kidneys to produce diuresis and natriuresis[118–120]. The diuretic effect of the exogenous BK administered by the renal artery is by B2 receptors, whereas the BK–induced natriuresis and increase in renal blood flow are due to both B1 and B2. Tissue injury, such as myocardial ischemia and inflammation, can cause the expression of B1 receptors on their surfaces[121]. Tryptase causes vascular permeability augmentation from Hageman (factor XII)-deficient plasma, whereas only 30% of this activity is produced from PK-deficient plasma [Table 1] [122]. Interestingly, no increase in vascular permeability occurred in kininogen-deficient plasma, suggesting the importance of kininogen in inflammatory diseases. An issue of particular interest is the question regarding the lack of complete abolishment of vascular permeability enhancement from PK-deficient plasma. Possible explanations for this observation may be due to heterozygosity of the donor cell genome or the presence of a tryptase-independent mechanism. Thus, the inhibitors of plasma kallikrein and BK receptors might be useful as therapeutic agents.
Allergic reactions
The effect of non-immune-mediated stimuli on several proinflammatory bioactive peptide release such as histamine and kinins are being revisited by researchers. The KKS plays roles in allergy, inflammation, and renal medicine[123]. Although many substances can induce allergic symptoms, dust mite is a major cause of in indoor allergies. Of interest, the activation of the KKS by the house dust mite Dematophagoides farinae (Df) proteinase is described[124]. It is demonstrated that Df proteinase can cause an increase in vascular permeability by activating the kinin-generating cascade without the release of histamine[124]. It suggests that Df proteinase-induced KKS activation contributes to kinin production in allergic reaction. Bradykinin mediates inflammation in patients with asthma due its bronchoconstrictor property. It has been demonstrated that inhaled BK causes the induction of bronchoconstriction[125] with a concomitted reduction in exhaled nitric oxide levels in asthmatic patients. Of note, angiotensin converting enzyme (ACE) expression is reduced in the epithelium of asthmatic patients[126]. ACE inactivates a number of peptide mediators, including BK. If ACE levels are down-regulated, BK levels are higher. The reduced ACE activity amplifies and sustains a large flux of local BK accumulation arising from the plasma KKS activation. A bradykinin B2 receptor antagonist (NPC 17731) blocked the antigen-induced asthmatic response in the primate and guinea pigs [127,128]. Similarly, a dose-dependent improvement in objective pulmonary function test occurred in adult patients treated with icatibant (HOE140, a selective bradykinin B2 receptor antagonist)[129]. Thus, the activation of KKS leading to increased BK formation is a physiological response which becomes pathophysiological in patients with asthma and related pathological conditions. For further details, see information about the physiology and molecular pharmacology of the KKS in [130–133].
Systemic lupus erthematosus
Systemic lupus erthematosus, generally referred to as lupus, is a chronic autoimmune disease in which the body’s antibodies attack one’s own tissues. Lupus attacks tissues such as the skin, muscles, tendons and ligaments as well as the kidneys, heart, lungs and brain. Lupus nephritis, inflammation of the kidney, occurs when antibodies and complement components build up in the kidneys and cause inflammation. It often causes nephrotic syndrome (excessive protein excretion) and may progress to renal failure, whereby nitrogen waste products build up in the bloodstream[134]. Recent investigations have shown that patients with systemic lupus erthematosus, presented with increased plasma concentrations of kinin precursors and an increase in plasma kallikrein activity. There is additional evidence which suggest lupus is associated with an increase risk of thrombosis brought on by higher incidence of thrombophilic mutations such as factor V leiden and prothrombin mutations[135]. For this reason, a plasma kallikrein inhibitor may offer an acute treatment option in which inflammatory cytokines may be decreased through the actions of inhibiting the KKS.
Hereditary angioedema
The above disease states all involve a hyperactivation of the inflammatory system in which the inhibition of the KKS may have some acute therapeutic potential. Conversely, inhibition of the KKS, as a chronic treatment of certain thrombotic conditions, may not be optimal due to the potential long term effects on the fibrinolytic system and vascular tone by BK. Over the past few years companies have looked at plasma kallikrein inhibition for treatments ranging from HAE to Pancreatitis. However, few to date have made it to market. In 2009 Ecallantide (DX-88), a recombinant inhibitor of PK for the treatment of severe acute hereditary angioedema (HAE) attacks was recommended for approval [Table 2]. The product was developed under a collaboration between Dyax and Genzyme. Dyax is also conducting phase II clinical trials for the reduction of peri-operative blood loss and the need for transfusion in patients undergoing cardiopulmonary bypass in the course of coronary artery bypass graft (CABG) surgery [Table 2] [136]. Even with the successful approval of Ecallantide issues remain regarding anaphylactic reactions which may occur[137]. Most of the plasma kallikrein inhibitors have been stopped either in the preclinical stage or the biological stages for various reasons. Recently preclinical studies looking at small molecule inhibitors of plasma kallikrein demonstrated good potency and selectivity in a preclinical setting[138]. Preclinical studies looking at the effect of PKSI-527 have shown beneficial effects in an LPS rat model of DIC [Table 2] [139]. Given the reported success of developing such inhibitors, it would be interesting to see if there would be a beneficial clinical effect on such conditions as sepsis, CPB and lupus.
Adult respiratory distress syndrome (ARDS)
ARDS is a complex inflammatory response and is characterized by endothelial injury. The activation of human plasma KKS in ARDS has been reported[140]. In these studies, the ratio between functional plasma kallikrein and PK-kallikrein antigen as an index for PK activation was examined. PK activation was significantly higher in patients with ARDS than that of the healthy subjects, suggesting that plasma KKS contributes to the pathogensis of ARDS[140]. Of interest, recent studies suggest that the degree of plasma KKS protein activation and capillary leak occurring in patients with adult respiratory distress syndrome (ARDS) is independent of the degree of factor VII-tissue factor activation and disseminated intravascular coagulation[141,142]. The need for a more comprehensive understanding of the mechanism of action of the KKS and the beneficial role of its inhibitors in ARDS is required.
Pregnancy
The activation of the plasma KKS in pregnancy has been reported[143]. It has been suggested that the inhibition of KKS may be a risk factor for early gestation losses[144]. The plasma KKS may contribute to the pathogenesis of pregnancy-induced hypertension (preeclampsia). Preeclampsia is a multifactorial and is a major cause of maternal and fetal morbidity and mortality. Preeclampsia is associated with an imbalance of vasoconstrictor and vasodilator molecules leading to a decrease in blood supply to the placenta.
Kininogen levels are significantly lower in chorion laeve, fetal placenta and maternal placenta in woman with preeclampsia than those of tissues from normotensive pregnant women[145]. The implications of this observation is that the activation of KKS may contribute to the local production and over-consumption of kininogen, influencing the pathogenesis of preeclampsia. While clinical management of preeclampsia does not currently include inhibitors of the KKS, we propose that KKS as a target for anti-preeclampsia therapy whereby inhibitors of this system may benefit patients with preeclampsia.
Angina pectoris
Intracoronary thrombus formation is critically important in unstable angina pectoris. Patients with unstable angina pectoris have significant increase in the plasma KKS activity with a concomitant increased BK generation, pointing toward a role for the plasma KKS in the coronary events[146]. Further, no abnormalities in the plasma kallikrein inhibition or in the antithrombin III levels is detected in patients with unstable angina pectoris[147]. Patients with stable angina and postinfarction cardiosclerosis in conjunction with a low stress tolerance had a marked increase in kinin levels during rationed exercise[148]. Plasma kallikrein is also involved in induction of elastase release from neutrophils and conversion of prourokinase to urokinase to initiate fibrinolysis[35,37,149,150]. Thus, the activation of plasma KKS would influence both vascular tone and leukocyte function in patients with recurrent angina.
Sickle cell nephropathy
The characteristic of sickle cell nephropathy is primarily associated with proteinuria, which starts in childhood and may lead to end-stage kidney disease[151,152]. The KKS activation occurs in association with both acute and chronic phases of cellular injury. The emerging evidence indicates an important role for KKS in a wide variety of chronic renal diseases. Of interest, both plasma and renal KKS are not activated in patients with polycystic kidney disease[153]. Glomerular hyperperfusion and hypertrophy leads to a chronic sickle cell nephropathy. ACE inhibitors slow down the progression of renal failure in sickle cell nephropathy, suggesting that glomerular capillary hypertension may be a risk factor in sickle cell nephropathy[154]. The upregulation of angiotensin type 1 (AT1) receptor expression has been documented in obstructive uropathy, suggesting that the bradykinin receptor expression level may be compromised[154]. Since the role of KKS in sickle cell nephropathy is inconclusive, further investigations are required to assess its role in this human pathophysiology.
Cardiac and vascular surgery
Another potential target involving the coagulation and inflammatory pathways is cardiopulmonary bypass (CPB) surgery. This type of procedure has been shown to induce a systemic inflammatory response characterized by pathological hypotension, disseminated intravascular coagulation and edema primarily caused by BK enhanced microvascular permeability and not histamine microvascular permeability. Additionally, inflammatory cytokines and adhesion molecules are known to be elevated, as can be expected during such a procedure. Investigations have revealed that an increase in BK liberation, brought on by the activation of the KKS, may be a major contributor to the adverse events that can follow CPB surgery[155]. The use of anticoagulants and anti-inflammatory drugs, such as heparin and aprotinin, have been in use for years during CPB procedures to reduce capillary leakage, preserve vascular resistance and improved myocardial recovery following ischemia[155]. This makes sense as aprotinin inhibits both plasma kallikrein and plasmin which are mediators of the proinflammatory system[155,156]. While aprotinin is used during these surgical procedures, it exhibits the same limitations that it does in the treatment for sepsis. A potent selective inhibitor of plasma kallikrein may have some utility in its ability to decrease the liberation of BK while at the same time decreasing the activation of the contact system. Indeed, plasma kallikrein inhibitors have been developed which suggest this would be a viable alternative to the use of aprotinin.
Pulmonary embolism
Embolization of a deep venous thrombosis (DVT) to the pulmonary artery might be the result of an increase in thrombolysis and/or the activation of the plasma KKS. Activation of KKS leads to the generation of BK and tissue plasminogen activator (tPA), two factors involved in promoting smooth blood flow through the arterial system. Investigations have been performed to determine the activation of the plasma KKS in patients undergoing major maxillofacial surgery and with some malignant disorders in order to identify the markers of KKS in DVT [157]. The plasma levels of KKS were not significantly increased in the two patient groups. However, tPA levels were significantly higher in the group of malignant patients on the first postoperative day, suggesting the activation of the KKS [Fig. (1)]. The inhibitor of plasma kallikrein might benefit tumor patients. Of interest, large-dose aprotinin (a kallikrein inhibitor) is effective in reducing blood loss in major orthopedic surgery[158].
Hemodialysis
Several independent clinical investigators have described the activation of the coagulation, plasma kallikrein and complement pathways during clinical hemodialysis during the past three decades[159–161]. Anaphylactoid reactions due to the activation of the KKS leading to BK generation in patients on angiotensin converting enzyme inhibitor (ACEI) therapy and hemodialysis with negatively charged dialysis membranes have been reported[161,162]. The stimulation of the KKS has been attributed to a decrease in alpha 1-proteinase inhibitor and alpha 2-macroglobulin levels in patients with chronic kidney insufficiency maintained on hemodialysis[159]. These studies suggest that KKS plays a role in the pathophysiological process and inhibitors of the biological effects of BK may contribute to the diminishing burden of hemodialysis.
Angiotensin converting enzyme inhibitor therapy
Plasma KKS counteracts the activity of the renin-angiotensin system. The renin-angiotensin-aldosterone system (RAAS) regulates blood pressure and water and electrolyte balance [Fig. (1)][163,164]. Angiotensin converting enzyme (ACE) metabolizes angiotensin I into angiotensin II (Ang II). Ang II stimulates migration and proliferation of vascular smooth muscle cells, cell adhesion and inhibition of macrophage-foam cell formation[164–167]. Ang II elevates blood pressure by binding and activating the angiotensin receptor Type 1 (AT1). Ang II is known to induce plasminogen activator inhibitor-1 (PAI-1, a potent inhibitor of tPA) gene expression in astroglial cells in the rat brain [Fig. (1)][168,169]. Ang II infusion elevates plasma PAI-1 and PAI-1 mRNA [170,171]. Ang II also triggers tissue factor production at the vascular level[172,173].
KKS modulates the activity of RAAS at several levels. Both gene targeting experiments in mice and computer simulation also point to the importance of interaction between KKS and RAAS[174]. ACE metabolizes BK, a major end-product of the KKS. Kallikrein converts prorenin to renin. PRCP (a PK activator), like angiotensin converting enzyme 2 (ACE2), metabolize Ang II (a vasoconstrictor, a prothrombotic risk factor) to angiotensin 1–7 (a vasodilator, an antithrombotic factor). Since ACE2 null mice do not exhibit elevated blood pressure compared to control littermates, PRCP may play a critical role in the control of normal vascular physiology. PRCP also converts angiotensin III to angiotensin 2–7. RAAS is considered to have a major role in promoting angiogenesis[175]. Since cleaved HK (HKa) is an antiangiogenic peptide, it is reasonable to suggest that HKa also modulates Ang II–induced neovascularization. Therefore, products of the KKS would influence the functions of RAAS.
Pemphigus Foliaceus
Pemphigus Foliaceus (PF) is normally a benign variety of pemphigus, an autoimmune blistering disease of both the skin and mucous membranes. The activation of both the intrinsic and extrinsic pathways of blood coagulation by PF has been examined. The expression of tissue factor and thrombomodulin on keratinocytes shielding with PF were found to be upregulated, indicating the activation of coagulation and sign of ongoing thrombosis [176]. The association of PF with HK levels and plasma kallikrein levels has been examined [177]. These studies demonstrated a reduction in HK levels but an increase in plasma kallikrein level in PF patients, indicating the activation of plasma KKS. However, an increased incidence in microthrombi generation or hypercoagulation in patients with PF does not appear to be a risk factor.
Emerging areas: more challenges
The complexity surrounding the kallikrein kinin system and its involvement in disease states is apparent. The difficulty is in determining whether the KKS provides a prothrombotic or an antithrombotic state. This will depend on understanding each disease state and at which stage the disease is in at the time of treatment. One can postulate that giving a plasma kallikrein inhibitor at the early stages of sepsis may not have the same effect as if it is given during the later stages, such as during septic shock. When considering plasma kallikrein inhibition, it is important to consider the major contributor factors (inflammation vs hemostasis), secondary contributing factors (coagulation/fibrinolysis), at what stage or time during the condition is plasma kallikrein most beneficial, and the time required for effectiveness. In using sepsis as an example, some considerations should be: When does the up-regulation of BK and its receptor B1 occur during this disease? Does the fibrinolytic process associated with plasma kallikrein and the contact play a significant role? How long would the inhibitor need to be on board and what risk is associated with the duration? Looking at plasma kallikrein inhibitors in this fashion highlights the possible therapeutic benefits, primarily in acute high inflammatory associated disease states.
Acknowledgments
This study was supported by NCRR/NIH P20RR021929 to SZ.
Footnotes
Conflict of Interest: The authors declare that they have no competing interests or financial or otherwise.
References
- 1.Hooley E, McEwan PA, Emsley J. J Thromb Haemost. 2007;5(12):2461. doi: 10.1111/j.1538-7836.2007.02792.x. [DOI] [PubMed] [Google Scholar]
- 2.Shariat-Madar Z, Schmaier AH. Trends Cardiovasc Med. 1999;9:238–44. doi: 10.1016/s1050-1738(00)00028-1. [DOI] [PubMed] [Google Scholar]
- 3.Tait JF, Fujikawa K. J Biol Chem. 1987;262:11651–56. [PubMed] [Google Scholar]
- 4.Thompson RE, Mandle R, Jr, Kaplan AP. J Clin Invest. 1977;60:1376–80. doi: 10.1172/JCI108898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colman RW, Schmaier AH. Crit Rev Oncol Hematol. 1986;5:57–85. doi: 10.1016/s1040-8428(86)80053-1. [DOI] [PubMed] [Google Scholar]
- 6.Griffin JH, Cochrane CG. Semin Thromb Hemost. 1979;5:254–73. doi: 10.1055/s-0028-1087158. [DOI] [PubMed] [Google Scholar]
- 7.Saito H. Adv Intern Med. 1980;25:217–38. [PubMed] [Google Scholar]
- 8.Schmaier AH, McCrae KR. J Thromb Haemost. 2007;5:2323–29. doi: 10.1111/j.1538-7836.2007.02770.x. [DOI] [PubMed] [Google Scholar]
- 9.Fong D, Chan MM, Hsieh WT. Biomed Biochim Acta. 1991;50:595–98. [PubMed] [Google Scholar]
- 10.Chung DW, Fujikawa K, McMullen BA, Davie EW. Biochemistry. 1986;25:2410–17. doi: 10.1021/bi00357a017. [DOI] [PubMed] [Google Scholar]
- 11.Colman RW, Schmaier AH. Blood. 1997;90:3819–43. [PubMed] [Google Scholar]
- 12.Asakura S, Yang W, Sottile J, Zhang Q, Jin Y, Ohkubo I, Sasaki M, Matsuda M, Hirata H, Mosher DF. J Biochem. 1998;124:473–84. doi: 10.1093/oxfordjournals.jbchem.a022138. [DOI] [PubMed] [Google Scholar]
- 13.Gustafson EJ, Schmaier AH, Wachtfogel YT, Kaufman N, Kucich U, Colman RW. J Clin Invest. 1989;84:28–35. doi: 10.1172/JCI114151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang YP, Muller-Esterl W, Schmaier AH. J Biol Chem. 1992;267:3712–17. [PubMed] [Google Scholar]
- 15.Colman RW. Adv Exp Med Biol. 1990;281:105–20. doi: 10.1007/978-1-4615-3806-6_11. [DOI] [PubMed] [Google Scholar]
- 16.Bano B, Kunapuli SP, Bradford HN, Colman RW. J Protein Chem. 1996;15:519–25. doi: 10.1007/BF01908533. [DOI] [PubMed] [Google Scholar]
- 17.Nieziolek M, Kot M, Pyka K, Mak P, Kozik A. Acta Biochim Pol. 2003;50:753–63. [PubMed] [Google Scholar]
- 18.Walsh PN. Semin Thromb Hemost. 2004;30:461–71. doi: 10.1055/s-2004-833481. [DOI] [PubMed] [Google Scholar]
- 19.Vogel R, Kaufmann J, Chung DW, Kellermann J, Muller-Esterl W. J Biol Chem. 1990;265:12494–502. [PubMed] [Google Scholar]
- 20.Weisel JW, Nagaswami C, Woodhead JL, La Cadena RA, Page JD, Colman RW. J Biol Chem. 1994;269:10100–06. [PubMed] [Google Scholar]
- 21.Pixley RA, Lin Y, Isordia-Salas I, Colman RW. J Thromb Haemost. 2003;1:1791–98. doi: 10.1046/j.1538-7836.2003.00291.x. [DOI] [PubMed] [Google Scholar]
- 22.Das A, Smalheiser NR, Markaryan A, Kaplan A. Biochim Biophys Acta. 2002;1571:225–38. doi: 10.1016/s0304-4165(02)00256-8. [DOI] [PubMed] [Google Scholar]
- 23.Colman RW. Curr Pharm Des. 2006;12:2599–607. doi: 10.2174/138161206777698710. [DOI] [PubMed] [Google Scholar]
- 24.Beaubien G, Rosinski-Chupin I, Mattei MG, Mbikay M, Chretien M, Seidah NG. Biochemistry. 1991;30:1628–35. doi: 10.1021/bi00220a027. [DOI] [PubMed] [Google Scholar]
- 25.Ciechanowicz A, Bader M, Wagner J, Ganten D. Biochem Biophys Res Commun. 1993;197:1370–76. doi: 10.1006/bbrc.1993.2628. [DOI] [PubMed] [Google Scholar]
- 26.Mandle RJ, Colman RW, Kaplan AP. Proc Natl Acad Sci USA. 1976;73:4179–83. doi: 10.1073/pnas.73.11.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang J, Yu CL, Williams SR, Springman E, Jeffery D, Sprengeler PA, Estevez A, Sampang J, Shrader W, Spencer J, Young W, McGrath M, Katz BA. J Biol Chem. 2005;280:41077–89. doi: 10.1074/jbc.M506766200. [DOI] [PubMed] [Google Scholar]
- 28.Adam A, Albert A, Calay G, Closset J, Damas J, Franchimont P. Clin Chem. 1985;31:423–26. [PubMed] [Google Scholar]
- 29.Schmaier AH. Curr Opin Hematol. 2000;7:261–65. doi: 10.1097/00062752-200009000-00001. [DOI] [PubMed] [Google Scholar]
- 30.Toossi Z, Sedor JR, Mettler MA, Everson B, Young T, Ratnoff OD. Proc Natl Acad Sci USA. 1992;89:11969–72. doi: 10.1073/pnas.89.24.11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghebrehiwet B, Silverberg M, Kaplan AP. J Exp Med. 1981;153:665–76. doi: 10.1084/jem.153.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin XP, Zeng SY, Tian HH, Deng SX, Ren JF, Zheng YB, Li D, Li YJ, Liao DF, Chen SY. Clin Exp Pharmacol Physiol. 2008 [Google Scholar]
- 33.Phipps JA, Clermont AC, Sinha S, Chilcote TJ, Bursell SE, Feener EP. Hypertension. 2009 doi: 10.1161/HYPERTENSIONAHA.108.117663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Discipio RG. Immunology. 1982;45:587–95. [PMC free article] [PubMed] [Google Scholar]
- 35.Kaplan AP, Kay AB, Austen KF. J Exp Med. 1972;135:81–97. doi: 10.1084/jem.135.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wachtfogel YT, Kucich U, James HL, Scott CF, Schapira M, Zimmerman M, Cohen AB, Colman RW. J Clin Invest. 1983;72:1672–77. doi: 10.1172/JCI111126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ichinose A, Fujikawa K, Suyama T. J Biol Chem. 1986;261:3486–89. [PubMed] [Google Scholar]
- 38.Klin Med (Mosk) 2007;85:54–57. [PubMed] [Google Scholar]
- 39.Colman RW, Wong PY. Thromb Haemost. 1977;38:751–75. [PubMed] [Google Scholar]
- 40.Martinez-Brotons F, Oncins JR, Mestres J, Amargos V, Reynaldo C. Thromb Haemost. 1987;58:709–13. [PubMed] [Google Scholar]
- 41.Cerf M, Raidoo D, Fink E, Fritz H, Bhoola K. Immunopharmacology. 1999;44:75–80. doi: 10.1016/s0162-3109(99)00112-5. [DOI] [PubMed] [Google Scholar]
- 42.Davie EW, Ratnoff OD. Science. 1964;145:1310–12. doi: 10.1126/science.145.3638.1310. [DOI] [PubMed] [Google Scholar]
- 43.Furie B, Furie BC. Cell. 1988;53:505–18. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 44.Hoffman MM, Monroe DM. Curr Hematol Rep. 2005;4:391–96. [PubMed] [Google Scholar]
- 45.Gailani D, Renne T. Arterioscler Thromb Vasc Biol. 2007;27:2507–13. doi: 10.1161/ATVBAHA.107.155952. [DOI] [PubMed] [Google Scholar]
- 46.Yousef GM, Diamandis EP. Clin Biochem. 2003;36:443–52. doi: 10.1016/s0009-9120(03)00055-9. [DOI] [PubMed] [Google Scholar]
- 47.Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Fifth. Lippincott William and Wilkins; 2006. [Google Scholar]
- 48.Schapira M, Scott CF, Colman RW. J Clin Invest. 1982;69:462–68. doi: 10.1172/JCI110470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colman RW. Immunopharmacology. 1999;43:103–08. doi: 10.1016/s0162-3109(99)00068-5. [DOI] [PubMed] [Google Scholar]
- 50.Coffman LG, Parsonage D, D’Agostino R, Jr, Torti FM, Torti SV. Proc Natl Acad Sci USA. 2009;106:570–75. doi: 10.1073/pnas.0812010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Colman RW. Semin Thromb Hemost. 2004;30:45–61. doi: 10.1055/s-2004-822970. [DOI] [PubMed] [Google Scholar]
- 52.Colman RW. Curr Pharm Des. 2006;12:2599–607. doi: 10.2174/138161206777698710. [DOI] [PubMed] [Google Scholar]
- 53.McCrae KR, Donate F, Merkulov S, Sun D, Qi X, Shaw DE. Curr Cancer Drug Targets. 2005;5:519–28. doi: 10.2174/156800905774574039. [DOI] [PubMed] [Google Scholar]
- 54.Xia CF, Yin H, Yao YY, Borlongan CV, Chao L, Chao J. Hum Gene Ther. 2006;17:206–19. doi: 10.1089/hum.2006.17.206. [DOI] [PubMed] [Google Scholar]
- 55.Zhang JC, Qi X, Juarez J, Plunkett M, Donate F, Sakthivel R, Mazar AP, McCrae KR. Can J Physiol Pharmacol. 2002;80:85–90. doi: 10.1139/y02-011. [DOI] [PubMed] [Google Scholar]
- 56.Cochrane CG, Revak SD, Wuepper KD. J Exp Med. 1973;138:1564–83. doi: 10.1084/jem.138.6.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cochrane CG, Griffin JH. Am J Med. 1979;67:657–64. doi: 10.1016/0002-9343(79)90253-5. [DOI] [PubMed] [Google Scholar]
- 58.Etscheid M, Beer N, Fink E, Seitz R, Johannes D. Biol Chem. 2002;383:1633–43. doi: 10.1515/BC.2002.184. [DOI] [PubMed] [Google Scholar]
- 59.Joseph K, Tholanikunnel BG, Kaplan AP. Int Immunopharmacol. 2002;2:1851–59. doi: 10.1016/s1567-5769(02)00186-8. [DOI] [PubMed] [Google Scholar]
- 60.Moreira CR, Schmaier AH, Mahdi F, da MG, Nader HB, Shariat-Madar Z. FEBS Lett. 2002;523:167–70. doi: 10.1016/s0014-5793(02)02980-0. [DOI] [PubMed] [Google Scholar]
- 61.Shariat-Madar Z, Mahdi F, Schmaier AH. Blood. 2004;103:4554–61. doi: 10.1182/blood-2003-07-2510. [DOI] [PubMed] [Google Scholar]
- 62.Shariat-Madar Z, Mahdi F, Schmaier AH. Int Immunopharmacol. 2002;2:1841–49. doi: 10.1016/s1567-5769(02)00178-9. [DOI] [PubMed] [Google Scholar]
- 63.Wuepper KD, Cochrane CG. J Exp Med. 1972;135:1–0. doi: 10.1084/jem.135.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mandle R, Jr, Kaplan AP. J Biol Chem. 1977;252:6097–104. [PubMed] [Google Scholar]
- 65.Rosing J, Tans G, Griffin JH. Eur J Biochem. 1985;151:531–38. doi: 10.1111/j.1432-1033.1985.tb09135.x. [DOI] [PubMed] [Google Scholar]
- 66.Patston PA, Gettins P, Beechem J, Schapira M. Biochemistry. 1991;30:8876–82. doi: 10.1021/bi00100a022. [DOI] [PubMed] [Google Scholar]
- 67.Guo YL, Colman RW. J Thromb Haemost. 2005;3:670–76. doi: 10.1111/j.1538-7836.2005.01218.x. [DOI] [PubMed] [Google Scholar]
- 68.Blaukat A. Andrologia. 2003;35:17–23. doi: 10.1046/j.1439-0272.2003.00533.x. [DOI] [PubMed] [Google Scholar]
- 69.Rhaleb, N. E.; Yang, X. P.; Nanba, M.; Shesely, E. G.; Carretero, O. A.
- 70.Kaplan AP, Joseph K, Shibayama Y, Nakazawa Y, Ghebrehiwet B, Reddigari S, Silverberg M. Clin Rev Allergy Immunol. 1998;16:403–29. doi: 10.1007/BF02737659. [DOI] [PubMed] [Google Scholar]
- 71.Zhao Y, Qiu Q, Mahdi F, Shariat-Madar Z, Rojkjaer R, Schmaier AH. Am J Physiol Heart Circ Physiol. 2001;280:H1821–H1829. doi: 10.1152/ajpheart.2001.280.4.H1821. [DOI] [PubMed] [Google Scholar]
- 72.Joseph K, Kaplan AP. Adv Immunol. 2005;86:159–208. doi: 10.1016/S0065-2776(04)86005-X. [DOI] [PubMed] [Google Scholar]
- 73.Sanden C, Enquist J, Bengtson SH, Herwald H, Leeb-Lundberg LM. J Pharmacol Exp Ther. 2008;326:24–32. doi: 10.1124/jpet.108.136911. [DOI] [PubMed] [Google Scholar]
- 74.Sheikh IA, Kaplan AP. Biochem Pharmacol. 1989;38:993–1000. doi: 10.1016/0006-2952(89)90290-6. [DOI] [PubMed] [Google Scholar]
- 75.Wennmalm A. J Intern Med. 1994;235:317–27. doi: 10.1111/j.1365-2796.1994.tb01081.x. [DOI] [PubMed] [Google Scholar]
- 76.Heffelfinger SC. Curr Pharm Des. 2007;13:1215–29. doi: 10.2174/138161207780618858. [DOI] [PubMed] [Google Scholar]
- 77.Souza DG, Lomez ES, Pinho V, Pesquero JB, Bader M, Pesquero JL, Teixeira MM. J Immunol. 2004;172:2542–48. doi: 10.4049/jimmunol.172.4.2542. [DOI] [PubMed] [Google Scholar]
- 78.Shariat-Madar Z, Mahdi F, Warnock M, Homeister JW, Srikanth S, Krijanovski Y, Murphey LJ, Jaffa AA, Schmaier AH. Blood. 2006;108:192–99. doi: 10.1182/blood-2006-01-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kimura S, Tsuji H, Nishimura H, Kato H, Ukimura N, Yano S, Kunieda Y, Kawano H, Nakagawa K, Nakagawa M. Thromb Res. 2002;106:41–50. doi: 10.1016/s0049-3848(02)00070-1. [DOI] [PubMed] [Google Scholar]
- 80.Witherow FN, Dawson P, Ludlam CA, Webb DJ, Fox KA, Newby DE. Arterioscler Thromb Vasc Biol. 2003;23:1667–70. doi: 10.1161/01.ATV.0000087142.99472.F6. [DOI] [PubMed] [Google Scholar]
- 81.Brown NJ, Gainer JV, Murphey LJ, Vaughan DE. Circulation. 2000;102:2190–96. doi: 10.1161/01.cir.102.18.2190. [DOI] [PubMed] [Google Scholar]
- 82.Stephens RW, Pollanen J, Tapiovaara H, Leung KC, Sim PS, Salonen EM, Ronne E, Behrendt N, Dano K, Vaheri A. J Cell Biol. 1989;108:1987–95. doi: 10.1083/jcb.108.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Behrendt N, List K, Andreasen PA, Dano K. Biochem J. 2003;371:277–87. doi: 10.1042/BJ20021508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schmaier AH, Silver L, Adams AL, Fischer GC, Munoz PC, Vroman L, Colman RW. Thromb Res. 1984;33:51–67. doi: 10.1016/0049-3848(84)90154-3. [DOI] [PubMed] [Google Scholar]
- 85.Vroman L. Nature. 1962;196:476–77. doi: 10.1038/196476a0. [DOI] [PubMed] [Google Scholar]
- 86.Weiss AS, Gallin JI, Kaplan AP. J Clin Invest. 1974;53:622–33. doi: 10.1172/JCI107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hathaway WE, Belhasen LP, Hathaway HS. Blood. 1965;26:521–32. [PubMed] [Google Scholar]
- 88.Katsuda I, Maruyama F, Ezaki K, Sawamura T, Ichihara Y. Eur J Haematol. 2007;79:59–68. doi: 10.1111/j.1600-0609.2007.00871.x. [DOI] [PubMed] [Google Scholar]
- 89.Francois D, Trigui N, Leterreux G, Flaujac C, Horellou MH, Mazaux L, Vignon D, Conard J, de MP. Blood Coagul Fibrinolysis. 2007;18:283–86. doi: 10.1097/MBC.0b013e328010bcde. [DOI] [PubMed] [Google Scholar]
- 90.Currimbhoy Z, Vinciguerra V, Palakavongs P, Kuslansky P, Degnan TJ. Am J Clin Pathol. 1976;65:970–74. doi: 10.1093/ajcp/65.6.970. [DOI] [PubMed] [Google Scholar]
- 91.LaDuca FM, Tourbaf KD. Am J Clin Pathol. 1981;75:626–28. doi: 10.1093/ajcp/75.4.626. [DOI] [PubMed] [Google Scholar]
- 92.Schmaier AH. J Clin Invest. 2002;109:1007–09. doi: 10.1172/JCI15490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pixley RA, La Cadena RA, Page JD, Kaufman N, Wyshock EG, Colman RW, Chang A, Taylor FB., Jr Am J Pathol. 1992;140:897–906. [PMC free article] [PubMed] [Google Scholar]
- 94.Annane D. Minerva Anestesiol. 2002;68:127–31. [PubMed] [Google Scholar]
- 95.Colman RW, Edelman R, Scott CF, Gilman RH. J Clin Invest. 1978;61:287–96. doi: 10.1172/JCI108938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De La Cadena RA, Laskin KJ, Pixley RA, Sartor RB, Schwab JH, Back N, Bedi GS, Fisher RS, Colman RW. Am J Physiol. 1991;260:G213–G219. doi: 10.1152/ajpgi.1991.260.2.G213. [DOI] [PubMed] [Google Scholar]
- 97.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Chest. 1992;101:1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 98.Riedemann NC, Guo RF, Ward PA. Nat Med. 2003;9:517–24. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 99.Robson W, Newell J. Nurs Stand. 2005;19:56–64. doi: 10.7748/ns2005.08.19.50.56.c3942. [DOI] [PubMed] [Google Scholar]
- 100.Sakkinen PA, Cushman M, Psaty BM, Kuller LH, Bajaj SP, Sabharwal AK, Boineau R, Macy E, Tracy RP. Thromb Haemost. 1998;80:134–39. [PubMed] [Google Scholar]
- 101.Herzum I, Renz H. Curr Med Chem. 2008;15:581–87. doi: 10.2174/092986708783769704. [DOI] [PubMed] [Google Scholar]
- 102.Russell JA. N Engl J Med. 2006;355:1699–713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 103.Aboab J, Nardi O, Lipiner D, Sharshar T, Annane D. Expert Opin Emerg Drugs. 2006;11:7–22. doi: 10.1517/14728214.11.1.7. [DOI] [PubMed] [Google Scholar]
- 104.Baue AE. J Trauma. 2003;55:997–98. doi: 10.1097/01.TA.0000094631.54198.07. [DOI] [PubMed] [Google Scholar]
- 105.Mattsson E, Herwald H, Cramer H, Persson K, Sjobring U, Bjorck L. Infect Immun. 2001;69:3877–82. doi: 10.1128/IAI.69.6.3877-3882.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marceau F. Immunopharmacology. 1995;30:1–26. doi: 10.1016/0162-3109(95)00011-h. [DOI] [PubMed] [Google Scholar]
- 107.Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, Calandra T, Dhainaut JF, Gerlach H, Harvey M, Marini JJ, Marshall J, Ranieri M, Ramsay G, Sevransky J, Thompson BT, Townsend S, Vender JS, Zimmerman JL, Vincent JL. Crit Care Med. 2008;36:296–327. doi: 10.1097/01.CCM.0000298158.12101.41. [DOI] [PubMed] [Google Scholar]
- 108.Fein AM, Bernard GR, Criner GJ, Fletcher EC, Good JT, Jr, Knaus WA, Levy H, Matuschak GM, Shanies HM, Taylor RW, Rodell TC. JAMA. 1997;277:482–87. [PubMed] [Google Scholar]
- 109.Otterbein L, Lowe VC, Kyle DJ, Noronha-Blob L. Agents Actions. 1993;39:C125–C127. doi: 10.1007/BF01972742. Spec No. [DOI] [PubMed] [Google Scholar]
- 110.Annane D, Bellissant E, Bollaert PE, Briegel J, Keh D, Kupfer Y. BMJ. 2004;329:480. doi: 10.1136/bmj.38181.482222.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang C, Qin J. Zhongguo Wuzhenxue Zazhi. 2008;8:1282–83. [Google Scholar]
- 112.Okajima K, Harada N. Curr Med Chem. 2006;13:2241–51. doi: 10.2174/092986706777935131. [DOI] [PubMed] [Google Scholar]
- 113.Hooper WC, Phillips DJ, Renshaw MA, Evatt BL, Benson JM. J Immunol. 1998;161:2567–73. [PubMed] [Google Scholar]
- 114.Gentry CA, Gross KB, Sud B, Drevets DA. Crit Care Med. 2009;37:19–25. doi: 10.1097/CCM.0b013e318192843b. [DOI] [PubMed] [Google Scholar]
- 115.Putterman C. Acta Chir Scand. 1989;155:367. [PubMed] [Google Scholar]
- 116.Dietrich W, Spath P, Ebell A, Richter JA. J Thorac Cardiovasc Surg. 1997;113:194–201. doi: 10.1016/S0022-5223(97)70415-X. [DOI] [PubMed] [Google Scholar]
- 117.Carretero OA, Scicli AG. Hypertension. 1991;18:I58–I69. doi: 10.1161/01.hyp.18.3_suppl.i58. [DOI] [PubMed] [Google Scholar]
- 118.Hulthen UL, Dymling JF, Hokfelt B. Acta Physiol Scand. 1980;110:307–14. doi: 10.1111/j.1748-1716.1980.tb06668.x. [DOI] [PubMed] [Google Scholar]
- 119.Sharma JN, Uma K, Noor AR, Rahman AR. Gen Pharmacol. 1996;27:55–63. doi: 10.1016/0306-3623(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 120.Yoshida H, Taniguchi S, Ura N. Nippon Rinsho. 2006;64(Suppl 2):199–202. [PubMed] [Google Scholar]
- 121.Lagneux C, Bader M, Pesquero JB, Demenge P, Ribuot C. Int Immunopharmacol. 2002;2:815–22. doi: 10.1016/s1567-5769(02)00022-x. [DOI] [PubMed] [Google Scholar]
- 122.Imamura T, Dubin A, Moore W, Tanaka R, Travis J. Lab Invest. 1996;74:861–70. [PubMed] [Google Scholar]
- 123.Fedoseev GB, Zhikharev SS, Goncharova VA, Kachanova TA, Razumovskaia TL. Ter Arkh. 1992;64:47–53. [PubMed] [Google Scholar]
- 124.Maruo K, Akaike T, Matsumura Y, Kohmoto S, Inada Y, Ono T, Arao T, Maeda H. Biochim Biophys Acta. 1991;1074:62–68. doi: 10.1016/0304-4165(91)90040-n. [DOI] [PubMed] [Google Scholar]
- 125.Kharitonov SA, Sapienza MM, Chung KF, Barnes PJ. Eur Respir J. 1999;14:1023–27. doi: 10.1183/09031936.99.14510239. [DOI] [PubMed] [Google Scholar]
- 126.Roisman GL, Danel CJ, Lacronique JG, henc-Gelas F, Dusser DJ. J Allergy Clin Immunol. 1999;104:402–10. doi: 10.1016/s0091-6749(99)70385-4. [DOI] [PubMed] [Google Scholar]
- 127.Ikemura T, Sasaki Y, Ohmori K. Clin Exp Allergy. 1998;28:635–43. doi: 10.1046/j.1365-2222.1998.00264.x. [DOI] [PubMed] [Google Scholar]
- 128.Hogan MB, Harris KE, Protter AA, Patterson R. Proc Assoc Am Physicians. 1997;109:269–74. [PubMed] [Google Scholar]
- 129.Akbary AM, Wirth KJ, Scholkens BA. Immunopharmacology. 1996;33:238–42. doi: 10.1016/0162-3109(96)00065-3. [DOI] [PubMed] [Google Scholar]
- 130.Kaplan AP. Postgrad Med. 1983;74:209–22. doi: 10.1080/00325481.1983.11698427. [DOI] [PubMed] [Google Scholar]
- 131.Marceau F, Bachvarov DR. Clin Rev Allergy Immunol. 1998;16:385–401. doi: 10.1007/BF02737658. [DOI] [PubMed] [Google Scholar]
- 132.Rodi D, Couture R, Ongali B, Simonato M. Curr Pharm Des. 2005;11:1313–26. doi: 10.2174/1381612053507422. [DOI] [PubMed] [Google Scholar]
- 133.Trifilieff A, Gies JP. Rev Mal Respir. 1991;8:339–50. [PubMed] [Google Scholar]
- 134.lalibera-Joviliano R, Reis ML, Donadi EA. Int Immunopharmacol. 2001;1:1889–96. doi: 10.1016/s1567-5769(01)00109-6. [DOI] [PubMed] [Google Scholar]
- 135.Pamuk GE, Turgut B, Vural O, Demir M, Soy M, Bozkurt G, Celik H. Clin Rheumatol. 2003;22:336–38. doi: 10.1007/s10067-003-0728-z. [DOI] [PubMed] [Google Scholar]
- 136.Lehmann A. Expert Opin Biol Ther. 2008;8:1187–99. doi: 10.1517/14712598.8.8.1187. [DOI] [PubMed] [Google Scholar]
- 137.Caballero T, Lopez-Serrano C. J Allergy Clin Immunol. 2006;117:476–77. doi: 10.1016/j.jaci.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 138.Young WB, Rai R, Shrader WD, Burgess-Henry J, Hu H, Elrod KC, Sprengeler PA, Katz BA, Sukbuntherng J, Mordenti J. Bioorg Med Chem Lett. 2006;16:2034–36. doi: 10.1016/j.bmcl.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 139.Katsuura Y, Okamoto S, Ohno N, Wanaka K. Thromb Res. 1996;82:361–68. doi: 10.1016/0049-3848(96)00085-0. [DOI] [PubMed] [Google Scholar]
- 140.Schapira M, Gardaz JP, Py P, Lew PD, Perrin LH, Suter PM. Bull Eur Physiopathol Respir. 1985;21:237–41. [PubMed] [Google Scholar]
- 141.Carvalho AC, DeMarinis S, Scott CF, Silver LD, Schmaier AH, Colman RW. J Lab Clin Med. 1988;112:270–77. [PubMed] [Google Scholar]
- 142.Fuhrer G, Heller W, Junginger W, Grober O, Roth K. Prog Clin Biol Res. 1989;308:737–42. [PubMed] [Google Scholar]
- 143.Strizhakov AN, Bol’shakova TD, Kuznetsov VA, Starkova TG. Akush Ginekol (Mosk) 1989:33–37. [PubMed] [Google Scholar]
- 144.Sugi T, Makino T. Am J Reprod Immunol. 2002;47:283–88. doi: 10.1034/j.1600-0897.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 145.Mohamed M, Larmie ET, Singh HJ, Othman MS. Eur J Obstet Gynecol Reprod Biol. 2006 doi: 10.1016/j.ejogrb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 146.Hoffmeister HM, Jur M, Wendel HP, Heller W, Seipel L. Circulation. 1995;91:2520–27. doi: 10.1161/01.cir.91.10.2520. [DOI] [PubMed] [Google Scholar]
- 147.Hoffmeister HM, Beyer ME, Engel Z, Heller W. Clin Cardiol. 1994;17:27–30. doi: 10.1002/clc.4960170106. [DOI] [PubMed] [Google Scholar]
- 148.Vinnikova MG, Makarevich I. Kardiologiia. 1987;27:56–59. [PubMed] [Google Scholar]
- 149.Discipio RG. Immunology. 1982;45:587–95. [PMC free article] [PubMed] [Google Scholar]
- 150.Wachtfogel YT, Kucich U, James HL, Scott CF, Schapira M, Zimmerman M, Cohen AB, Colman RW. J Clin Invest. 1983;72:1672–77. doi: 10.1172/JCI111126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bergmann S, Zheng D, Barredo J, Abboud MR, Jaffa AA. J Pediatr Hematol Oncol. 2006;28:147–53. doi: 10.1097/01.mph.0000203722.91189.9d. [DOI] [PubMed] [Google Scholar]
- 152.Schwartz GJ, Lerner NB. J Pediatr Hematol Oncol. 2006;28:111–14. doi: 10.1097/01.mph.0000203718.37824.2e. [DOI] [PubMed] [Google Scholar]
- 153.Braun C, Kleemann T, Birck R, Hilgenfeldt U, Riester U, Tschope C, van der Woude FJ, Rohmeiss P. Int Immunopharmacol. 2002;2:1949–56. doi: 10.1016/s1567-5769(02)00171-6. [DOI] [PubMed] [Google Scholar]
- 154.Harris RC, Martinez-Maldonado M. Miner Electrolyte Metab. 1995;21:328–35. [PubMed] [Google Scholar]
- 155.Mojcik CF, Levy JH. Ann Thorac Surg. 2001;71:745–54. doi: 10.1016/s0003-4975(00)02218-9. [DOI] [PubMed] [Google Scholar]
- 156.Campbell DJ, Dixon B, Kladis A, Kemme M, Santamaria JD. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1059–R1070. doi: 10.1152/ajpregu.2001.281.4.R1059. [DOI] [PubMed] [Google Scholar]
- 157.Wendel HP, Scholpp J, Schulze HJ, Heller W, Schwenzer N. J Craniomaxillofac Surg. 1999;27:266–70. doi: 10.1016/s1010-5182(99)80039-0. [DOI] [PubMed] [Google Scholar]
- 158.Samama CM, Langeron O, Rosencher N, Capdevila X, Rouche P, Pegoix M, Berniere J, Coriat P. Anesth Analg. 2002;95:287–93. doi: 10.1097/00000539-200208000-00005. table. [DOI] [PubMed] [Google Scholar]
- 159.Shcherbakova EG, Varakina NI, Iarovaia GA, Belorusov OS, Dotsenko VL, Vorob’eva LS. Vopr Med Khim. 1991;37:40–43. [PubMed] [Google Scholar]
- 160.Schulman G, Hakim R, Arias R, Silverberg M, Kaplan AP, Arbeit L. J Am Soc Nephrol. 1993;3:1563–69. doi: 10.1681/ASN.V391563. [DOI] [PubMed] [Google Scholar]
- 161.Krieter DH, Steinke J, Kerkhoff M, Fink E, Lemke HD, Zingler C, Muller GA, Schuff-Werner P. Artif Organs. 2005;29:47–52. doi: 10.1111/j.1525-1594.2004.29007.x. [DOI] [PubMed] [Google Scholar]
- 162.Amore A, Guarnieri G, Atti M, Schena FP, Coppo R. J Nephrol. 1999;12:383–89. [PubMed] [Google Scholar]
- 163.Farmer JA, Torre-Amione G. Curr Atheroscler Rep. 2001;3:117–24. doi: 10.1007/s11883-001-0047-2. [DOI] [PubMed] [Google Scholar]
- 164.Campbell DJ. Int J Biochem Cell Biol. 2003;35:784–91. doi: 10.1016/s1357-2725(02)00262-5. [DOI] [PubMed] [Google Scholar]
- 165.De MW. Regul Pept. 2003;114:87–90. doi: 10.1016/s0167-0115(03)00121-6. [DOI] [PubMed] [Google Scholar]
- 166.Baltatu O, Bader M. Neuroendocrinology. 2003;78:253–59. doi: 10.1159/000074446. [DOI] [PubMed] [Google Scholar]
- 167.Buchner N, Vonend O, Rump LC. Herz. 2006;31:294–302. doi: 10.1007/s00059-006-2821-y. [DOI] [PubMed] [Google Scholar]
- 168.Lottermoser K, Weisser B, Hertfelder HJ, Wostmann B, Vetter H, Dusing R. Am J Hypertens. 1998;11:378–84. doi: 10.1016/s0895-7061(97)00496-2. [DOI] [PubMed] [Google Scholar]
- 169.Zelezna B, Rydzewski B, Lu D, Olson JA, Shiverick KT, Tang W, Sumners C, Raizada MK. Mol Endocrinol. 1992;6:2009–17. doi: 10.1210/mend.6.12.1491687. [DOI] [PubMed] [Google Scholar]
- 170.Skurk T, Lee YM, Hauner H. Hypertension. 2001;37:1336–40. doi: 10.1161/01.hyp.37.5.1336. [DOI] [PubMed] [Google Scholar]
- 171.Raizada MK, Lu D, Yang H, Yu K. Adv Exp Med Biol. 1996;396:71–78. doi: 10.1007/978-1-4899-1376-0_8. [DOI] [PubMed] [Google Scholar]
- 172.Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, Speth RC, Raizada MK, Katovich MJ. Am J Physiol Heart Circ Physiol. 2007;292:H736–H742. doi: 10.1152/ajpheart.00937.2006. [DOI] [PubMed] [Google Scholar]
- 173.Dechend R, Muller DN, Wallukat G, Homuth V, Krause M, Dudenhausen J, Luft FC. Semin Nephrol. 2004;24:571–79. doi: 10.1016/s0270-9295(04)00128-7. [DOI] [PubMed] [Google Scholar]
- 174.Katsuya T, Ogihara T. Nippon Rinsho. 2004;62:111–19. [PubMed] [Google Scholar]
- 175.Nagai N, Oike Y, Izumi-Nagai K, Koto T, Satofuka S, Shinoda H, Noda K, Ozawa Y, Inoue M, Tsubota K, Ishida S. Invest Ophthalmol Vis Sci. 2007;48:2321–26. doi: 10.1167/iovs.06-1296. [DOI] [PubMed] [Google Scholar]
- 176.Mizutani H, Ohyanagi S, Nouchi N, Inachi S, Shimizu M. Australas J Dermatol. 1996;37(Suppl 1):S48–S49. doi: 10.1111/j.1440-0960.1996.tb01085.x. [DOI] [PubMed] [Google Scholar]
- 177.Rosatelli TB, Roselino AM, lalibera-Joviliano R, Reis ML, Donadi EA. Br J Dermatol. 2005;152:650–57. doi: 10.1111/j.1365-2133.2005.06427.x. [DOI] [PubMed] [Google Scholar]
- 178.Damas J. Peptides. 1996;17:859–72. doi: 10.1016/0196-9781(96)00056-3. [DOI] [PubMed] [Google Scholar]
- 179.Isordia-Salas I, Pixley RA, Li F, Sainz I, Balfour SR, Adam A, Colman RW. Int Immunopharmacol. 2002;2:1895–905. doi: 10.1016/s1567-5769(02)00183-2. [DOI] [PubMed] [Google Scholar]
- 180.Kaschina E, Stoll M, Sommerfeld M, Steckelings UM, Kreutz R, Unger T. Physiol Genomics. 2004;19:41–49. doi: 10.1152/physiolgenomics.00035.2004. [DOI] [PubMed] [Google Scholar]
- 181.Koch M, Bonaventura K, Spillmann F, Dendorfer A, Schultheiss HP, Tschope C. Biol Chem. 2008;389:719–23. doi: 10.1515/BC.2008.083. [DOI] [PubMed] [Google Scholar]
- 182.Kouyoumdjian M, Damas J. Arch Physiol Biochem. 1998;106:25–32. doi: 10.1076/apab.106.1.25.4400. [DOI] [PubMed] [Google Scholar]
- 183.Oh-Ishi S. Nippon Yakurigaku Zasshi. 1993;101:209–18. doi: 10.1254/fpj.101.4_209. [DOI] [PubMed] [Google Scholar]
- 184.Oh-Ishi S, Hayashi I, Yamaki K, Utsunomiya I. Agents Actions Suppl. 1992;38(Pt 1):277–91. doi: 10.1007/978-3-0348-7321-5_36. [DOI] [PubMed] [Google Scholar]
- 185.Oh-Ishi S, Hayashi I, Satoh K, Nakano T. Thromb Res. 1984;33:371–77. doi: 10.1016/0049-3848(84)90076-8. [DOI] [PubMed] [Google Scholar]
- 186.Colman RW, Bradford HN, Warner AH. Thromb Res. 1989;54:115–23. doi: 10.1016/0049-3848(89)90041-8. [DOI] [PubMed] [Google Scholar]
- 187.Cheung PP, Kunapuli SP, Scott CF, Wachtfogel YT, Colman RW. J Biol Chem. 1993;268:23361–65. [PubMed] [Google Scholar]
- 188.Krijanovski Y, Proulle V, Mahdi F, Dreyfus M, Muller-Esterl W, Schmaier AH. Blood. 2003;101:4430–36. doi: 10.1182/blood-2002-11-3329. [DOI] [PubMed] [Google Scholar]
- 189.Colman RW. Braz J Med Biol Res. 1994;27:1839–53. [PubMed] [Google Scholar]
- 190.Exner T, Barber S, Naujalis J. Med J Aust. 1987;146:545–47. doi: 10.5694/j.1326-5377.1987.tb120397.x. [DOI] [PubMed] [Google Scholar]
- 191.Gallimore MJ, Harris SL, Jones DW, Winter M. Thromb Res. 2004;114:91–96. doi: 10.1016/j.thromres.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 192.Matheson RT, Miller DR, Lacombe MJ, Han YN, Iwanaga S, Kato H, Wuepper KD. J Clin Invest. 1976;58:1395–406. doi: 10.1172/JCI108595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hayashi H, Ishimaru F, Fujita T, Tsurumi N, Tsuda T, Kimura I. Rinsho Ketsueki. 1989;30:1694–701. [PubMed] [Google Scholar]
- 194.Hayashi H, Ishimaru F, Fujita T, Tsurumi N, Tsuda T, Kimura I. Blood. 1990;75:1296–304. [PubMed] [Google Scholar]
- 195.Ishimaru F, Dansako H, Nakase K, Fujii N, Sezaki N, Nakayama H, Fujii N, Komiyama Y, Iijima K, Takenaka K, Teshima T, Shinagawa K, Ikeda K, Niiya K, Harada M. Int J Hematol. 1999;69:126–28. [PubMed] [Google Scholar]
- 196.Iijima K, Nakamura K. Rinsho Ketsueki. 1989;30:1702–07. [PubMed] [Google Scholar]
- 197.Lefrere JJ, Horellou MH, Gozin D, Conard J, Muller JY, Clark M, Soulier JP, Samama M. Am J Hematol. 1986;22:415–19. doi: 10.1002/ajh.2830220411. [DOI] [PubMed] [Google Scholar]
- 198.Raja SN, Campbell JN, Meyer RA, Colman RW. Clin Sci (Lond) 1992;83:337–41. doi: 10.1042/cs0830337. [DOI] [PubMed] [Google Scholar]
- 199.Hayashi H, Koya H, Kitajima K, Kimura I. Acta Med Okayama. 1978;32:81–83. [PubMed] [Google Scholar]
- 200.Asmis LM, Sulzer I, Furlan M, Lammle B. Thromb Res. 2002;105:463–70. doi: 10.1016/s0049-3848(02)00045-2. [DOI] [PubMed] [Google Scholar]
- 201.Saito H, Ratnoff OD. Adv Exp Med Biol. 1979;120B:61–70. [PubMed] [Google Scholar]
- 202.Donaldson VH, Kleniewski J, Saito H, Sayed JK. J Clin Invest. 1977;60:571–83. doi: 10.1172/JCI108809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lombardi AM, Sartori MT, Cabrio L, Fadin M, Zanon E, Girolami A. Thromb Haemost. 2003;90:1040–45. doi: 10.1160/TH03-05-0275. [DOI] [PubMed] [Google Scholar]
- 204.Scicli AG, Mindroiu T, Scicli G, Carretero OA. J Lab Clin Med. 1982;100:81–93. [PubMed] [Google Scholar]
- 205.Wuillemin WA, Furlan M, von Felten A, Lammle B. Thromb Haemost. 1993;70:427–32. [PubMed] [Google Scholar]
- 206.Saito H, Goldsmith GH., Jr Blood. 1977;50:377–85. [PubMed] [Google Scholar]
- 207.Stern DM, Nossel HL, Owen J. J Clin Invest. 1982;69:1270–76. doi: 10.1172/JCI110566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kravtsov DV, Wu W, Meijers JC, Sun MF, Blinder MA, Dang TP, Wang H, Gailani D. Blood. 2004;104:128–34. doi: 10.1182/blood-2003-10-3530. [DOI] [PubMed] [Google Scholar]
- 209.Poon MC, Moore MR, Castleberry RP, Lurie A, Huang ST, Lehmeyer J. Am J Hematol. 1982;12:261–70. doi: 10.1002/ajh.2830120308. [DOI] [PubMed] [Google Scholar]
- 210.Iwaki T, Castellino FJ. Thromb Haemost. 2006;95:1003–10. doi: 10.1160/TH06-03-0128. [DOI] [PubMed] [Google Scholar]
- 211.Lodi S, Isa L, Pollini E, Bravo AF, Scalvini A. Scand J Haematol. 1984;33:80–82. doi: 10.1111/j.1600-0609.1984.tb02214.x. [DOI] [PubMed] [Google Scholar]
- 212.Patrassi GM, Martinelli S, Vianello C, Girolami A. Folia Haematol Int Mag Klin Morphol Blutforsch. 1982;109:644–54. [PubMed] [Google Scholar]
- 213.Chhibber G, Cohen A, Lane S, Farber A, Meloni FJ, Schmaier AH. J Lab Clin Med. 1990;115:112–21. [PubMed] [Google Scholar]
- 214.Cugno M, Cicardi M, Bottasso B, Coppola R, Paonessa R, Mannucci PM, Agostoni A. Blood. 1997;89:3213–18. [PubMed] [Google Scholar]
- 215.Lammle B, Zuraw BL, Heeb MJ, Schwarz HP, Berrettini M, Curd JG, Griffin JH. Thromb Haemost. 1988;59:151–61. [PubMed] [Google Scholar]
- 216.Cugno M, Hack CE, de Boer JP, Eerenberg AJ, Agostoni A, Cicardi M. J Lab Clin Med. 1993;121:38–43. [PubMed] [Google Scholar]
- 217.Davis AE. Clin Immunol. 2005;114:3–9. doi: 10.1016/j.clim.2004.05.007. [DOI] [PubMed] [Google Scholar]