

Abstract
The functional and 13C isotopic compositions of water-soluble organic carbon (WSOC) in atmospheric aerosol were determined by nuclear magnetic resonance (1H-NMR) and isotope ratio mass spectrometry (IRMS) in an urban location in the Southern Mississippi Valley. The origin of WSOC was resolved using the functional distribution of organic hydrogen, δ13C ratio, and positive matrix factorization (PMF). Three factors were retained based on NMR spectral bins loadings. Two factors (factors 1 and 3) demonstrated strong associations with the aliphatic region in the NMR spectra and levoglucosan resonances. Differences between the two factors included the abundance of the aromatic functional group for factor 1, indicating fresh emissions and, for factor 3, the presence of resonances attributed to secondary ammonium nitrate and low δ13C ratio values that are indicative of secondary organic aerosol. Factors 1 and 3 added 0.89 and 1.08 μgC m−3, respectively, with the highest contribution in the summer and fall. Factor 2 retained resonances consistent with saccharides and was attributed to pollen particles. Its contribution to WSOC varied from 0.22 μgC m−3 in winter to 1.04 μgC m−3 in spring.
Keywords: Organic aerosol, Biomass burning, NMR, Secondary organic aerosol
Graphical Abstract
1. Introduction
The composition and size distribution of atmospheric aerosol play a critical role in microphysical processes and cloud formation (Shen et al., 2012; Lin et al., 2014; Ruehl and Wilson, 2014). Organic carbon (OC) is a major component of aerosol contributing up to 90% of aerosol mass with the water-soluble fraction of organic carbon (WSOC) being responsible for 20–70% of OC (Park et al., 2003; Stone et al., 2008; Hand et al., 2012). WSOC is directly involved in cloud condensation nuclei (CCN) by modifying the aqueous chemistry and surface tension of cloud droplets (Mircea et al., 2002; Ernvens et al., 2005; Shen et al., 2012). Despite its significance, very little is known on WSOC chemical composition and its sources, with less than 10–20% of organic mass being structurally identified (Cappiello et al., 2003; Ding et al., 2008; Chan et al., 2013; Villalobos et al., 2013).
WSOC is composed of a mixture of high-molecular-weight carboxylic, keto/carbonyl, amino/imino, and nitro multifunctional organic compounds, and is frequently referred to as humic-like substances (HULIS) as well as smaller organic molecules, such as anhydrides, sugars and keto- and α,ω-dicarboxylic acids (Graham et al., 2002; Cappiello et al., 2003; Fuzzi et al., 2006; Duarte et al., 2007; Ding et al., 2008). An approach to determine WSOC compositional characteristics involved the identification of functional groups using proton nuclear magnetic resonance (1H-NMR). NMR is a non-destructive method that provides information on the different types of non-exchangeable organic hydrogen in solution (Chalbot and Kavouras, 2014). NMR analysis was performed on different types of organic aerosol (OA), namely, biomass burning aerosol, marine organic aerosol, secondary organic aerosol (SOA), urban aerosol, rural aerosol and size fractionated atmospheric aerosol (Graham et al., 2002; Duarte et al., 2007; Decesari et al., 2001, 2005, 2011; Chalbot et al., 2014).
The molecular markers approach has been extensively applied to reconcile OA sources, but, it cannot provide quantitative estimates on source contributions. Receptor modeling including positive matrix factorization (PMF) has been applied to identify and quantify WSOC sources/types (Heo et al., 2013; Paglione et al., 2014). PMF analyzes the variability in multiple measurements to extract a limited number of factors and attribute them to specific sources/types based on loadings. While PMF and other factorial-based receptor modeling methods have limitations associated with reactivity of individual chemical species and statistical power, they have successfully been applied to reconcile the sources of organic compounds.
Fine particle mass in the Southern Mississippi Valley was composed of secondary sulfate and nitrate (about 46%) and smoke particles (about 25%). Wood burning is extensively used for domestic heating in the winter, while prescribed fires, wildfires and recreational open fires are frequent in spring and summer. Traffic exhaust and diesel combustion accounted for 12.5% (Chalbot et al., 2013a). The chemical analysis of size distributed organic aerosol in the same area in winter and spring further confirmed the dominant contribution of biomass burning emissions and secondary inorganic aerosol on fine WSOC (Chalbot et al., 2014). Traces of traffic exhaust and secondary organic aerosol tracers were observed in the ultrafine range, and biological particles in the coarse fraction (with diameter > 3.0 μm). The air quality in this region was also influenced by sources in the Gulf of Mexico, Texas, Southeast U.S., Great Plains and Pacific Northwest depending on the season (Lee and Hopke, 2006; Chalbot et al., 2013a). According to the 2011 U.S. Environmental Protection Agency’s National Emission Inventory, 52.5% (75,015 tons) of PM2.5 emissions were from fires and biomass burning in Arkansas. Mobile sources and domestic/industrial activities using fossil fuels added a merely 7340 tons of PM2.5 (or 5% of PM2.5 emissions). It is noteworthy that these estimates may not include recreational log and leaf fires in backyards, a widespread activity in Southern Mississippi. This fact is evidenced by the number of warnings to not burn tornado debris issued by the Arkansas Department of Environmental Quality and Health following the EF5 tornado in Vilonia, AR in April 2015.
The goal of this study was to apportion WSOC of atmospheric aerosol collected in the Southern Mississippi Valley. The specific objectives of this study were: (i) to identify the functional composition of WSOC by 1H-NMR, thermal optical reflectance (TOR) and isotope ratio mass spectrometry; (ii) to determine its seasonal variability; and (iii) to identify and quantify its types/sources. PMF was employed to identify the most important OA sources and quantify their contribution to WSOC by multivariate linear regression analysis.
2. Materials and methods
2.1. Sample collection and preparation
A total of fifty-one 3-day urban total suspended aerosol (TSP) samples were collected every other week on a periodic schedule (e.g. sample #1 was collected on Monday–Tuesday–Wednesday, sample #2 was collected on Tuesday–Wednesday–Thursday) in Little Rock, Arkansas from December 2012 to November 2014. The frequency and duration of sampling was selected to reduce the effect of sampling biases (i.e., weekday/weekend) and obtain sufficient quantities for concurrent NMR, isotopic 13C, and TOR analysis. The sampling site was located at the northeast corner of the University of Arkansas for Medical Sciences campus. A GMWL-2000 sampler was used (Tisch Environmental, Ohio, USA). Particles were collected on pre-baked 203 × 254 mm quartz fiber filter (QM-A grade, Tisch Environmental, USA), having collection efficiency > 99.999% for particles with radius larger than 0.3 μm at an operating flow rate of 1.13 m3 min−1. Particle mass (PM) determination and extraction methodology has been described elsewhere (Chalbot et al., 2014). Briefly, one half of each filter was extracted by sonication with 50 ml of H2O for 1 h. The total water soluble extract (TWSE) was concentrated under rotary evaporation transferred to a pre-weighed vial and dried using a SpeedVac and stored in a freezer at −30 °C until analysis.
2.2. WSOC and 13C analysis
An aliquot of the extract was analyzed for WSOC using a DRI Model 2001 TOR Carbon Analyzer (Atmoslytic Inc., Calabasas, CA) at Desert Research Institute Environmental Analysis Facility (Ho et al., 2006; Chalbot et al., 2014). WSOC concentrations are expressed in μgC m−3. δ13C was measured on the water-soluble extract by a coupled elemental analyzer/isotope ratio mass spectrometer (NC2500 Carlo Erba/Delta Plus) at the University of Arkansas Stable Isotope Laboratory (Nelson, 2000). Data was normalized using two in-house standards (acetanilide −31.81, benzoic acid −27.77) and one certified reference standard (IAEA-C6-10.45). Data are expressed in permil (‰) versus the Vienna Pee Dee Belemnite (VPDB) using a combination of certified and in house standards.
2.3. 1H-NMR spectroscopy
All samples were analyzed on a Bruker Avance 500 MHz spectrometer equipped with a 5 mm BBFO Plus Smart Probe operated at 298 K. The samples were reconstituted in D2O containing trimethylsilyl-propionic acid-d4 sodium salt (TSP-d4; 0.248 μmol/ml) as the internal standard (IS) for the analyses. NMR spectra were manually corrected, aligned and integrated using the ACD/NMR processor (Version 12.01 Academic Edition). The integration of NMR spectra was completed twice. First, resonance signals in five pre-defined ranges were integrated as follows: (i) H–C (δH 0.6–1.8 ppm) that included R–CH3, R–CH2, and R–CH protons; (ii) H–C–C= (δH 1.8–3.2 ppm) which included allylic protons; (iii) H–C–O (δH 3.2–4.4 ppm) contained protons contiguous to alcohols, ethers and ester functionalities. Protons in secondary and tertiary amines may also appear in this range; (v) O–CH–O and H–C= (δH 5.0–6.4 ppm) included protons of carbohydrates, olefins, ethers, esters and organic nitrates; and (iv) H–Ar (δH 6.5–8.3 ppm) contained aromatic protons (Decesari et al., 2001; Chalbot et al., 2014). Note that other types on organic hydrogen may appear in these pre-defined regions (Chalbot and Kavouras, 2014). The boundaries and definitions of the ranges were consistent with those previously used in examining the functional composition of organic aerosol by NMR and to facilitate direct comparison with prior studies (Decesari et al., 2001). Secondly, the intensity of signals in 250 pre-defined bins with 0.04 ppm width was integrated for the PMF analysis. Since the signal intensity is proportional to the number of protons, the non-exchangeable organic hydrogen concentrations were calculated based on the known concentrations of the internal standard (i.e. TSP) (Bharti and Roy, 2012). The region δH 4.5–5.0 ppm was eliminated due the presence of the residual water resonance. The region δH 7.0–7.5 ppm, due to inclusion of resonances of ammonium, was also eliminated from WSOC reconstruction and PMF analysis.
2.4. Materials and quality assurance
The high volume sampler was calibrated every six months per manufacturer’s instructions. Filters were placed in clean glass containers, wrapped with aluminum foil and stored in a freezer at −30 °C until extraction. Pure solvents (HPLC grade) and standard compounds (Fisher Scientific) were used for extraction and identification of compounds. Glassware used in extraction and analysis was pre-cleaned at 450 °C for 12 h. Filter blanks were analyzed using the same method as the samples (Pal et al., 2015). Sample shipment met the requirements of the EC/OC analysis protocol to reduce the risk of contamination (DRI, 2001).
2.5. Carbon reconstruction and PMF analysis
The WSOC concentration associated with each type of non-exchangeable organic hydrogen was computed by the following linear model (Chalbot et al., 2014):
| (1) |
where α1–5 were the regression coefficient to convert the non-exchangeable organic hydrogen concentrations for each type to μgC m−3, αo was the intercept (i.e. WSOC associated with other types of organic hydrogen such as O=C–O–H, C–O–H and tertiary C). Categorical “design” variables (True = 1; False = 0) were added to determine the effect of season (regression coefficients: α6 through α9). The regression coefficients α1–5 were the combinations of contributions of each functional group to WSOC and the molar H/C ratio for each organic hydrogen group. The goodness of the fit was assessed by examining the χ2 and the Akaike Information Criterion (AIC). The robustness of the regression analysis was assessed using the Coefficient of Variation of the relative squared mean error between measured and reconstructed WSOC (CVRMSE).
The underlying assumption in PMF is that the concentration (xij) on i-day at the j-receptor site is the linear sum of the products of source contributions (gik) and profiles (fkj) of k-sources through the minimization of the objective function (Q in Eq. (2)) and imposing non-negative constraints (Paatero and Tapper, 1994; Paatero et al., 2002):
| (2) |
where σij is the uncertainty of xij and hij is a filter function to handle outliers in the dataset
| (3) |
where α is the user-defined outlier threshold distance.
The US Environmental Protection Agency PMF (version 5.0) model was applied to apportion WSOC sources using the 250 NMR bins (US EPA, 2014). The uncertainty matrix was calculated using the relative standard deviation of reference NMR spectra and a coverage factor (k) of 2 (95% confidence interval). There were no missing concentration or uncertainty values. Of the fifty-six (56) NMR bins with signal-to-noise (S/N) ratio (calculated based on measured concentrations and uncertainties by PMF) less than 0.5 were eliminated from PMF analysis. Fifteen (15) NMR bins with 0.5 < S/N < 1.0 were categorized as weak and their corresponding uncertainty increased by a factor of three. An additional uncertainty of 15% was added to reduce modeling errors. The base run was executed with the assigned factors ranging from 2 to 8. The configuration included 100 runs with random seed per each run. All base run results were converged. The bootstrap configuration included 500 runs, minimum r value of 0.75 and suggested block of 4 with a random seeding. The inter-quantile ranges for individual compounds were ±5. Runs with Fpeak values from −2 to +2 with an increment of 0.1 did not improve the solution. The contributions of the retained factors to the WSOC concentrations were computed using a least-squares multivariate linear regression model
| (4) |
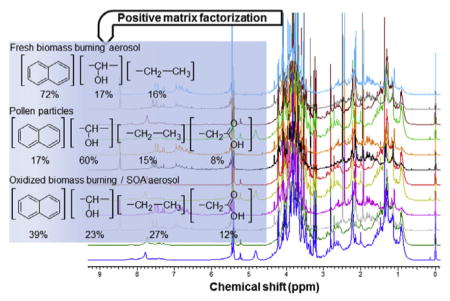
where βk is the regression coefficient or scaling constant to convert the factor profiles and contributions to μgC m−3 (Song et al., 2001; Vargas et al., 2014). The intercept, α, was attributed to other WSOC sources. Negative regression coefficients indicated that too many factors were used in the regression. The optimum number of factors (sources) and the rotation (controlled by the Fpeak value) was selected using both statistical metrics and assessing the robustness and physical meaning of the regression results (Paatero et al., 2002, 2005). For the three factor solution, Q remained relatively constant, the highest individual column mean (IM) and standard deviation (IS) from the scaled residual matrix dropped significantly and the highest element in rotmat increased. Solutions with more than 6 factors were eliminated due to negative contribution to WSOC, while solutions with 4 and 5 factors were excluded due to strong correlations between factors and poor/unstable contributions to WSOC. The profiles and contributions of PMF models with 2,3,4 and 5 factors are presented in the Supporting information (Figs. S1–S5). The profiles of retained factors were compared against 1H-NMR spectral signatures of vegetation combustion, pollen and dust particles (Chalbot et al., 2013a,b).
3. Results and discussion
3.1. Functional characteristics
Fig. 1 shows the typical 1H-NMR spectra of the WSOC for each trimester (March–April–May (MAM, spring); June–July–August (JJA, summer); September–October–November (SON, fall); December–January–February (DJF, winter)) and the five regions for different types of non-exchangeable organic hydrogen. Table 1 shows the minimum, maximum, mean and standard deviation of PM, TWSE, WSOC, total and per type non-exchangeable organic hydrogen concentrations, the δ13C isotopic and molar H/C ratios in each seasonal trimester.
Fig. 1.

Typical 500 MHz 1H-NMR spectra of WSOC in fall (SON), winter (DJF), spring (MAM) and summer (JJA). (TSP-d4: Internal Standard, Fo: Formate, L: Levoglucosan, G: glucose, S: Sucrose, Su: Succinate, A: Acetate, F: Fructose, T: Trigonelline, PA: Phthalate and TPA: Terephthalate, Phe, Phenylalanine, Tyr, Tyrosine, Ala: Alanine, Ile: Isoleucine, Leu: Leucine, Val: Valine, MMA: Monomethylamine, DMA, Dimethylamine, TMA: Trimethylamine, MSA: Methanesulfonate).
Table 1.
Particle Mass (PM), Total Water Soluble Extract (TWSE), WSOC, Non-Exchangeable Organic Hydrogen Concentrations, δ13C, and molar H/C ratios (min – max; mean (standard deviation)) in aerosol samples.
| Parameter | Winter (SON) N = 14
|
Spring (MAM) N = 12
|
Summer (JJA) N = 12
|
Fall (SON) N = 13
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | σ | Min | Max | Mean | σ | Min | Max | Mean | σ | Min | Max | Mean | σ | Min | Max | |
| TSP (μg/m3) | 25.9 | 13.2 | 11.7 | 59.7 | 41.1 | 17.7 | 25.2 | 73.9 | 35.2 | 8.7 | 19.2 | 50.3 | 38.8 | 36.5 | 18.4 | 158.1 |
| TWSE (μg/m3) | 8.2 | 2.9 | 4.4 | 13.7 | 15.2 | 16.9 | 5.0 | 67.9 | 12.3 | 2.9 | 6.9 | 17.2 | 8.8 | 3.9 | 0.1 | 14.8 |
| WSOC (μg/m3) | 1.8 | 1.0 | 0.7 | 4.0 | 2.7 | 0.7 | 1.6 | 3.9 | 3.4 | 1.3 | 1.7 | 5.9 | 2.5 | 0.8 | 1.4 | 3.8 |
| Org. H (nmol/m3) | 58.8 | 29 | 27.5 | 130.7 | 98.3 | 27.5 | 44.7 | 153.6 | 130.5 | 68.0 | 56.4 | 275.4 | 90.1 | 34.5 | 38.7 | 133.3 |
| H–C | 21.4 | 11.9 | 8.7 | 55.5 | 29.1 | 7.6 | 16.6 | 43.9 | 48.9 | 26.6 | 20.9 | 104.7 | 32.0 | 13.3 | 13.7 | 49.2 |
| H–C–C= | 17.6 | 8.9 | 7.8 | 39.1 | 24.4 | 6.3 | 13.2 | 38.4 | 36.0 | 22.9 | 12.5 | 82.4 | 26.0 | 11.9 | 11.6 | 40.5 |
| H–C–O | 17.1 | 9.3 | 8.0 | 40.0 | 40.8 | 22.8 | 12.5 | 85.7 | 41.2 | 18.1 | 19.6 | 80.9 | 28.0 | 11.4 | 9.5 | 47.5 |
| O–CH–O & H–C= | 1.7 | 0.8 | 0.7 | 3.2 | 2.7 | 1.1 | 1.3 | 5.1 | 2.6 | 1.0 | 1.6 | 4.6 | 2.3 | 0.8 | 0.8 | 3.3 |
| H–Ar | 1.1 | 0.5 | 0.3 | 1.8 | 1.2 | 0.5 | 0.5 | 2.3 | 1.7 | 1.0 | 0.6 | 3.7 | 1.8 | 0.9 | 0.6 | 3.5 |
| δ13C ratio (‰) | −26.20 | −25.03 | −26.55 | −24.82 | −27.20 | −24.76 | −25.95 | −23.90 | ||||||||
The average (standard deviation in parentheses) total suspended particle (TSP) mass varied from 25.9 (13.2) μg m−3 in winter to 41.1 (17.7) μg m−3 in spring with the highest sample PM concentration (158.1 μg m−3) measured in fall. The mean TWSE concentrations ranged from 8.2 (2.9) μg m−3 in winter to 15.2 (16.9) μg m−3 in spring. A different seasonal pattern was observed for WSOC and non-exchangeable organic hydrogen with the highest mean concentrations measured in the summer, 3.4 (1.3) μgC m−3 and 130.46 (68.04) ng m−3, respectively. For fall, winter and summer WSOC, H–C was the predominant type of non-exchangeable organic hydrogen (from 21.41 to 48.87 ng m−3) followed by H–C–C= (17.55–36.04 ng m−3) and H–C–O (17.06–41.18 ng m−3) (Table 1). On the other hand, a strong carbohydrate signature was computed for aerosol samples collected in spring with H–C–O representing about 40% of total non-exchangeable organic hydrogen (Table 1) due to the presence of pollen-dominated organic aerosol (Chalbot et al., 2013b, 2014). The strong H–C–O signature during summer may be associated with SOA formation and aged organic aerosol (Cristofanelli et al., 2013). Very little variability was observed for the H–Ar and O–CH–O/H–C=fractions.
The NMR spectra were described by sharp resonances of the most abundant organic species and overlapping spectral resonances of many organic compounds. Certain resonances were assigned to specific organic compounds using reference NMR spectra and in comparison with previous studies (Wishart et al., 2009; Chalbot et al., 2014). These included low molecular weight acids [acetate (at δ 1.92 ppm), succinate (at δ 2.41 ppm) and formate (δ 8.47 ppm)] associated with both primary and secondary OA; amines (at δ 2.59, δ 2.72, and δ 2.92 ppm) and (at δ 7.0–7.4 ppm), associated with secondary inorganic aerosol, levoglucosan (at δ 3.52, δ 3.67, δ 3.73–3.75, δ 4.08 ppm and δ 5.45 ppm) associated with biomass burning emissions, sugars [glucose (δ 3.24, δ 3.37–3.43 ppm, δ 3.44–3.49, δ 3.52, δ 3.68–3.73, δ 3.74–3.77, δ 3.88–3.91, δ 3.81–3.85 and δ 5.23 ppm), fructose (at δ 3.46, δ 3.55, δ 3.67, δ 3.75, δ 3.82, δ 3.87, δ 3.89, δ 4.06, δ 4.22 and δ 5.41 ppm) and sucrose (δ 3.55–3.61, δ 3.66–3.73, δ 3.79–3.84, δ 3.89–3.91, δ 3.99–4.04 and δ 4.11–4.12 ppm)]; and tracers of anthropogenic [phthalic acid (δ 7.58 and δ 7.73 ppm), terephthalic acid (δ 8.01 ppm] and marine [methanesulfonic acid at δ 3.55–3.61 ppm] SOA.
The identification of these compounds indicated the presence of a mixture of OA sources. Smoke particles were present throughout the year and secondary, inorganic and organic, aerosol in winter and summer were associated with both anthropogenic and marine precursors were mostly observed in the summer, which was associated with the highest prevalence of resonances of oxygenated organic species.
3.2. WSOC reconstruction
The attribution of WSOC to specific carbon associated with the four types of non-exchangeable organic hydrogen was done with the multivariate regression analysis described in Section 2.4 Equation (1). Fig. 2 shows the agreement between the measured and estimated WSOC for each season (Figure S6 shows the percentage contributions). The five types of WSOC 1H NMR resonances explained most of measured WSOC (unassigned: from 0.1 to 1.58 μgC m−3) with CVRMSE of 0.04 (or 4%) with most of the unassigned WSOC in winter. This may be a result of organic carbon associated with protons not identified by this NMR experimental configuration. These include exchangeable protons found in carboxylic, alcoholic and phenolic groups.
Fig. 2.
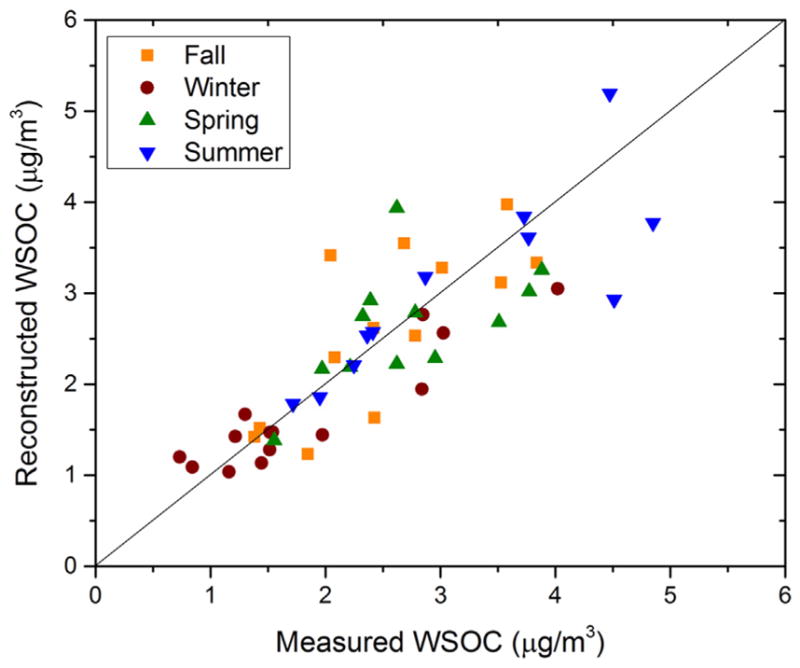
Measured and reconstructed WSOC (by linear regression) in urban aerosol.
Fig. 3 depicts the contribution of specific types of carbon using the H–C, H–C–C=, H–C–O, O–CH–O/H–C= and H–Ar definition of non-exchangeable organic hydrogen to WSOC for difference seasons (based on the regression results). The H–Ar carbon was the most abundant WSOC type in fall (26%) and winter (22%) followed by H–C–O (23% and 20% in fall and winter, respectively) and H–C (21% and 20%, respectively) carbon. The high content of aromatic carbon hinted the potential contribution of combustion sources. On the other hand, WSOC illustrated different compositional characteristics in spring and summer with the H–C–O carbon being the dominant WSOC contributor (32% and 26% in spring and summer), followed by H–C (19% and 25%, respectively) and H–C–C= (16% and 19%, respectively) carbons. This pattern may be attributed to the contribution of pollen and other biogenic particles in spring with a strong carbohydrate signature. Oxygenated organic compounds may also be formed through the reactions of organic compounds with OH radicals and ozone in summer resulting in SOA formation and/or atmospheric aging of primary organic aerosol (Shakya et al., 2012; Cristofanelli et al., 2013).
Fig. 3.
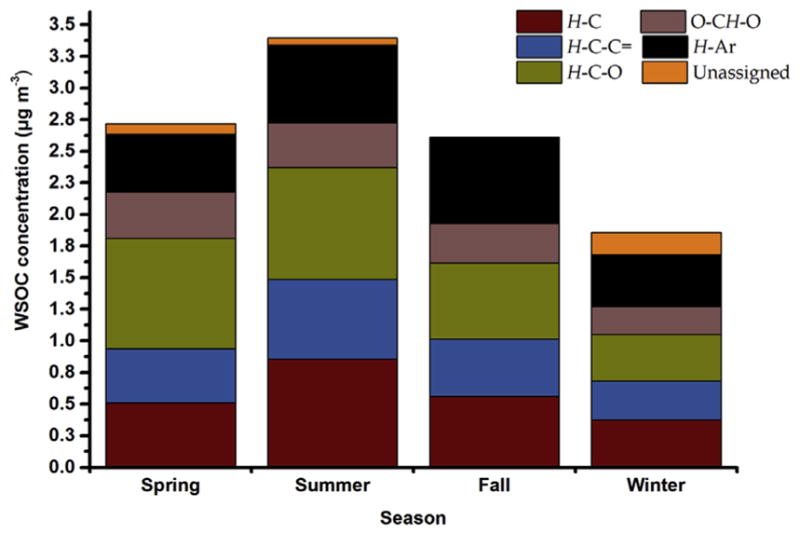
Seasonal contributions of H–C, H–C–C=, H–C–O, O–CH–O and H–Ar to WSOC in urban aerosol.
3.3. Functional group distribution and δ13C ratio
Fig. 4 shows the distribution of WSOC functional groups of aerosol samples in Southern Mississippi Valley for each trimester and published boundaries of SOA, marine OA, biomass burning, urban aerosol and biogenic particles (Decesari et al., 2006, 2011; Shakya et al., 2012; Cleveland et al., 2012). The WSOC samples in the Southern Mississippi Valley were characterized by H–C–C=O/sum(aliphatics) ratio values from 0.20 to 0.45 and H–C–O//sum(aliphatics) ratio values from 0.31 to 0.68. Relatively high H–C–O and H–C–C=O and elevated H–Ar contents were measured for most of the samples collected in winter and spring and to a lesser extent in spring and summer. None of the samples were within the marine and SOA boxes (red and blue boxes in Fig. 4), nor did they fall into the urban aerosol range (black trapezoid in Fig. 4) (Cleveland et al., 2012). A subset of samples had H–C–C=O/sum(aliphatics) and H–C–O//sum(aliphatics) ratio values that were comparable to those in rural aerosol in the Northeast US (Shakya et al., 2012). A number of samples were within the boundaries of aerosol collected downwind of biomass burning emission in Rondônia, Brazil (Graham et al., 2002; Decesari et al., 2006), Mediterranean Sea aerosol (Cristofanelli et al., 2013) and wood burning in winter, both fine and ultrafine WSOC in the Southern Mississippi Valley (Chalbot et al., 2014). However, a subset of samples were outside the boundaries of the five OA types probably due to the polyols and anhydrides associated with fresh pollen and smoke particles (with high H–C–O but low H–C–C=O contributions). For spring samples, the H–C–C=O and H–C–O contents were comparable to the H–C–C=O/sum(aliphatics) and H–C–O//sum(aliphatics) ratio values computed for pollen-dominated coarse particles in the same region (Chalbot et al., 2014). The remaining samples had resonances previously attributed to levoglucosan, a tracer of wood burning. These samples were also characterized by declining H–C–C=O/sum(aliphatics) and increasing H–C–O/sum(aliphatics) ratios, which may be characteristic of primary emissions. Note that the aerosol samples from Rondônia, Brazil and the Mediterranean Sea were collected downwind of biomass burning. Further, atmospheric aging of primary biomass burning emissions progressed as expected by the reduced prevalence of polyols and phenols (Graham et al., 2002; Decesari et al., 2006; Cristofanelli et al., 2013). Thus, samples within the biomass burning region (blue box in Fig. 4) may represent aged biomass burning aerosol while those outside may be associated with fresh emissions.
Fig. 4.

Functional group distribution of WSOC in the Southern Mississippi Valley.
The δ13C ratio values per sample varied from −27.20‰ in the summer to −23.90‰ in fall. While, on average, there were no substantial differences of δ13C ratio values among the four seasons, a smaller range (1.17) was computed for winter as compared to the other seasons (1.41–2.15). These values were comparable to those measured for size-distributed WSOC (−26.81‰ to −25.93‰) and within the range of δ13C ratio values for C3 plants (i.e. plants that fix inorganic CO2 using the C3 carbon fixation metabolic pathway), unleaded gasoline and diesel exhaust (Collister et al., 1994; Widory et al., 2004; Chalbot et al., 2014). The lowest δ13C ratio values may also be indicative of SOA formation through the oxidation of volatile organic compounds. However, atmospheric aging processes result in δ13C ratio increase (Aggarwal et al., 2013).
3.4. Source apportionment
This section presents the apportionment of WSOC using the integrated 1H-NMR spectra in 250 bins. The number of extracted factors was set based on PMF statistical outputs and WSOC regression analysis. Our results were robust to sensitivity tests including randomly selected subsets and perturbation datasets, since loadings and estimates were altered by less than 5%. As mentioned, the region δH 7.0–7.5 ppm was eliminated from PMF analysis. However, since ammonium nitrate and/or sulfate are tracers of secondary aerosol and regional transport in the Southern Mississippi Valley, we determined the factor profiles (but did not consider factor contributions) with and without the N–H resonances. Fig. 5 shows the profiles of the three retained factors for WSOC with and without NH4 + resonances. The very good consistency of factor profiles for the two runs further demonstrated the stability of factor profiles and provided useful information on WSOC type and origin. Minor differences (less than 2%) for factors 1 and 3 were associated with the loadings of individual NMR bins. This may be related to associations of NMR resonances of organic compounds such as methylamines (co-emitted from husbandry facilities) with (Chalbot et al., 2014).
Fig. 5.

Profiles for the three retained factors at the Southern Mississippi Valley for PMF run with and without resonances.
The contribution of each factor to WSOC for each sample and the δ13C is presented in Fig. 6. In addition, the mean seasonal contribution and functional composition of each factor to WSOC concentration is depicted in Fig. 7a and b, respectively. The contribution of factor loading to WSOC was computed using the regression analysis model described in Equation (4), while the functional composition for each factor was computed using the factor profiles and converting the non-exchangeable organic hydrogen concentrations associated with each factor to WSOC using pre-defined molar H/C ratios of 2 for the aliphatic saturated and usaturated, 1.1. for the carbonhydrate and oxygenated fractions, and 0.4 for the aromatic fraction (Decesari et al., 2007). The calculated WSOC mass concentration was found to be ~5% less than the corresponding measured WSOC concentration. This suggested that the majority of WSOC was contributed by the three factors. The CVRSME was 0.14 (14%). The profiles of factors 1 and 3 were comparable in the H–C, H–C–C= and H–C–O ranges, exhibiting strong humic-like substances and anhydrides NMR spectral signatures (Fig. 5). Both factors also retained substantial associations with the region containing resonances attributed to levoglucosan (at δH ~5.5. ppm) (Fig. 5). These patterns were comparable to those previously observed for HULIS in different environments (Graham et al., 2002; Decesari et al., 2006). However, the two factors demonstrated differences in the H–Ar fraction with stronger associations of H–Ar bins with factor 1 than with factor 3 (Fig. 7b). Ammonium resonances were retained by factor 3, while broad NMR resonances were retained in factor 1. Ammonium nitrate was previously observed in fine and ultrafine atmospheric aerosol in the Southern Mississippi Valley during winter and originated from regional sources along the Midwest and Pacific Northwest (Chalbot et al., 2013c). This indicated that both factors may be associated with a specific type of organic aerosol, namely, biomass burning (with the additional contribution of secondary aerosol, including nitrate, for factor 3). Factors 1 and 3 contributed 0.89 and 1.08 μgC m−3, respectively, with comparable seasonal profiles and the highest contributions in summer and fall (Figs. 6 and 7a). Factor 1 was dominated by H–Ar and H–C carbon that may indicate fresh combustion emissions (Fig. 7b). On the contrary, the saturated and unsaturated aliphatic carbon accounted for most of factor 3’s WSOC followed by H–C–O which is consistent with the presence of oxygenated compounds from SOA formation and/or oxidized biomass burning aerosol (Cristofanelli et al., 2013; Paglione et al., 2014).
Fig. 6.

WSOC, δ13C ratio and contributions of the three retained PMF factors for individual urban aerosol samples.
Fig. 7.

(a) Seasonal contributions of the three retained factors to WSOC; and (b) functional composition of the three retained factors.
We previously determined that biomass burning contributed 3.0 μg/m3 to PM2.5; however, the profiles of the remaining factors also contained elemental and organic carbon (Chalbot et al., 2013a,b,c,d). The outcomes of the PMF analysis were re-processed by regressing the factor contributions to organic carbon (OC) and elemental carbon (EC) levels to estimate the influence of biomass burning on PM2.5 OC and EC. Biomass burning added 41% and 62% of OC and EC respectively. Regional sources including secondary inorganic (organic sea salt aerosol) and contaminated sea salt contributed 28% and 15%, respectively. Fossil fuel combustion including primary traffic exhaust and diesel inputs were accounted for 6% for OC and 3% for EC. About 20% of both OC and EC were not accounted for by the retained sources.
To obtain additional insight into the origin of carbonaceous aerosol during sampling days, levels of PM2.5 elemental carbon, organic carbon, soluble potassium (K+) and total potassium (K) measured at the nearest U.S. EPA NCore site (EPA AIRS ID: 051190007; 34°45′21.9″N and 92° 15′4.1″W; sampling frequency 1-in-3 days) were retrieved. The site is located 3.6 km ENE (heading of 77.9°) from our sampling site. The TSP WSOC levels correlated very well with PM2.5 OC, EC and K+ for individual samples indicating common origin (Fig. S5). Soluble K+ is a tracer of biomass burning (Andreae, 1983). OC/EC ratios less than two are indicative of primary traffic emissions, while biomass burning and SOA formation result in OC/EC values higher than four (Turpin and Lim, 2001). It is worth noting that samples with TSP WSOC higher than PM2.5 OC suggest the presence of coarse WSOC that is usually of biological origin (Chalbot et al., 2014).
Table 2 shows the mean (±st.error) of OC/EC and K+/K diagnostic rations for the Dec.2012–Nov. 2014 period, for days corresponding to sample days in this study and for samples with the 15% highest contributions of the three retained factors. In agreement with the 2002–2012 period, OC/EC and K+/K ratios for the period 2012–2014 were 6.51 ± 0.37 and 0.86 ± 0.08, demonstrating the dominant influence of biomass burning as compared to fossil fuel combustion sources. The values of both ratios for NCore samples corresponding to samples obtained in this study were comparable to those observed for the period 2012–2014. The K+/K and OC/EC values for factor 1 (0.93 ± 0.16 and 5.35 ± 0.73) were consistently higher than those observed in traffic-dominated urban areas indicating the predominant influence of biomass burning on factor 1. The higher OC/EC values were computed for days with the highest loadings for factor 3 (6.62 ± 1.10) but lower K+/K values as compared to those computed for Factor 1 may be due to SOA formation from biogenic and anthropogenic VOCs. For most of the days with high loadings for both factors 1 and 3, the δ13C varied from −26.5 to −25.0‰. For the aerosol samples collected in June 2013 (starting dates of June 10, 2013 and June 21, 2013), the δ13C values (lower than −27‰) (WSOC was associated with factor 3 and to a lesser extend with factor 1) indicated the possible influence of SOA formation.
Table 2.
Diagnostic ratios of PM2.5 chemical species during the sampling period and samples with high contributions of the three retained factors by PMF.
| OC/EC | K+/K | |
|---|---|---|
| 2012–2014 | 6.51 ± 0.37 | 0.86 ± 0.08 |
| Sampling days | 6.31 ± 0.29 | 0.70 ± 0.05 |
| Factor 1 | 5.35 ± 0.73 | 0.93 ± 0.16 |
| Factor 2 | 6.32 ± 0.70 | 0.61 ± 0.06 |
| Factor 3 | 6.62 ± 1.10 | 0.68 ± 0.07 |
In addition, distinct differences were observed by examining the origin of air masses arriving at Southern Mississippi Valley during days with high factor 1 and 3 contributions. The residence time of air masses for the samples with the 20% highest contributions of factors 1 and 3 to WSOC, respectively, were computed by calculating hourly air mass backward trajectories every one hour going back 5 days using National Oceanic Atmospheric Administration’s Hybrid Single Particle Trajectory (HYSPLIT) model. Trajectory analysis and residence time calculations were done using TrajStat, ArcMap and SPSS, as previously described (Chalbot et al., 2013c).
Fig. 8 shows the difference of air masses trajectory times (a), and the air mass backward trajectories and locations of wildfires for days with high factor 1 and 3 contributions (b and c, respectively). Negative and positive values in Fig. 8a show the origin of air mass with high contributions to factors 1 and 3, respectively. Both factors showed a combination of local and regional influences. The regional component of factor 1 was associated with transport from Eastern Texas and the Caribbean (Fig. 8a,b). For factor 3, regional contributions originated from the Upper Midwest and Pacific Northwest. These regions experienced intense fire events, albeit at different seasons. The frequency of fires in the Southeastern United States and Caribbean Islands peaked in spring and early summer, while fires in the Western United States were more frequent in late summer and spring (Chalbot et al., 2013d). Given the factors’s 3 profile and its seasonal variability, it may be attributed to mixed biomass burning emissions and secondary aerosol.
Fig. 8.

(a) Difference of residence time of air masses with high contribution of factors 1 and 3 in a horizontal grid cell using backward trajectories at 500 m above ground level (b) air mass backward trajectories and MODIS fire detection for samples with high contribution of Factor 1; and (c)) air mass backward trajectories and MODIS fire detection for samples with high contribution of Factor 3.
The profile of factor 2 showed strong associations with the H–C–O range and particularly, the NMR bins encompassing resonances attributable to saccharides such as glucose and fructose. Less pronounced associations with the H–C and H–C–C= ranges were computed. The factor profile was comparable to the 1H-NMR spectra of pollen particles obtained in the same region (Chalbot et al., 2014). Factor 2 added 0.52 μgC m−3 with the highest contribution (1.04 μgC m−3) in spring and the lowest in winter.
4. Conclusions
The composition of the particulate WSOM has been studied in the Southern Mississippi Valley. The types of WSOC were reconciled based on the types of non-exchangeable organic hydrogen resolved by NMR spectroscopy. Analysis of the WSOC functional composition and δ13C demonstrated the presence of oxygenated organic compounds. Smoke was detected due to the presence of levoglucosan and saturated aliphatic and carbohydrate signatures attributed to other biomass-related anhydrides. Pollen particles in the spring also demonstrated a strong saccharides-induced carbohydrate signature. Ammonium resonances in winter indicated the contribution of secondary inorganic aerosol, while MSA and phthalic acids revealed the formation of SOA from marine and anthropogenic precursors. The types of sources of WSOC were apportioned using PMF. Two of three retained factors exhibited strong similarities such as the presence of levoglucosan. Analysis of fine OC, EC and soluble K+ during the measurement period clearly demonstrated the dominant contribution of biomass burning on both factors. The presence of NMR resonances attributed to secondary components, high OC/EC ratios and δ13C ratios for factor 3 suggested the presence of SOA. The results from this study indicated that PMF analysis of WSOC’s NMR spectral patterns may provide unique insights into here the types and origins of WSOC as a whole and for specific functional groups such as H–C–O that govern cloud and fog formation. Future studies may examine the types of WSOC for different particle sizes in order to differentiate the signature of SOA to primary OA. The functional composition of fractionated WSOM (i.e. non-polar, polar and acidic fractions) may also provide insights into the chemical composition of different types of WSOM.
Supplementary Material
HIGHLIGHTS.
We characterized the water-soluble organic matter (WSOM) of ambient aerosol by NMR.
Biomass burning and secondary aerosol dominated WSOM in the summer.
Pollen particles accounted for most of WSOM in spring.
Acknowledgments
We would like thank Dr. Rebecca Helm and Allyn Holladay for editing the manuscript. This study was partially supported in part by The Deep South Center for Occupational Health and Safety (Grant # 5T42OH008436 from NIOSH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIOSH and the US Food and Drug Administration.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.atmosenv.2015.12.067.
References
- Aggarwal SG, Kawamura K, Umarji GS, Tachibana E, Patil RS, Gupta PK. Organic and inorganic markers and stable C-, N-isotopic compositions of tropical coastal aerosols from megacity Mumbai: sources of organic aerosols and atmospheric processing. Atmos Chem Phys. 2013;13:4667–4680. [Google Scholar]
- Andreae MO. Soot carbon and excess fine potassium: long-range transport of combustion-derived aerosols. Science. 1983;220:1148–1151. doi: 10.1126/science.220.4602.1148. [DOI] [PubMed] [Google Scholar]
- Bharty SK, Roy R. Quantitative 1H-NMR spectroscopy. Trends Anal Chem. 2012;35:5–26. [Google Scholar]
- Cappiello A, De Simoni E, Fiorucci C, Mangani F, Palma P, Trufelli H, Decesari S, Facchini MC, Fuzzi S. Molecular characterization of the water-soluble organic compounds in fogwater by ESIMS/MS. Environ Sci Technol. 2003;37(7):1229–1240. [Google Scholar]
- Chalbot MCG, Kavouras IG. Nuclear magnetic resonance spectroscopy for determining the functional content of organic aerosols: a review. Environ Poll. 2014;91:232–249. doi: 10.1016/j.envpol.2014.04.034. [DOI] [PubMed] [Google Scholar]
- Chalbot MG, da Costa GG, Kavouras IG. NMR analysis of the water-soluble fraction of airborne pollen particles. App Magn Res. 2013a;44:1347–1358. [Google Scholar]
- Chalbot MC, McElroy B, Kavouras IG. Sources, trends and regional impacts of fine particulate matter in southern Mississippi valley: significance of emissions from sources in the Gulf of Mexico coast. Atmos Chem Phys. 2013b;13:3721–3732. [Google Scholar]
- Chalbot MC, Nikolich G, Etyemezian V, Dubois DW, King J, Shafer D, da Costa GG, Hinton JF, Kavouras IG. Soil humic-like organic compounds in prescribed fire emissions using nuclear magnetic resonance spectroscopy. Environ Poll. 2013c;181:167–171. doi: 10.1016/j.envpol.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Chalbot MC, DuBois DW, Kavouras IG. Assessment of the contribution of wildfires on ozone concentrations in the central US-Mexico border region. Aer Air Qual Res. 2013d;13:838–848. [Google Scholar]
- Chalbot MG, Brown J, Chitranshi P, da Costa GG, Pollock ED, Kavouras IG. Functional characterization of the water-soluble organic carbon of size-fractionated aerosol in the southern Mississippi Valley. Atmos Chem Phys. 2014;14:6075–6088. doi: 10.5194/acp-14-6075-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AWH, Issacman G, Wilson KR, Worton DR, Ruehl CR, Nah T, Gentner DR, Dallmann TR, Kirchstetter TW, Harley RA, Gilman JB, Kuster WC, de Gouw JA, Offenberg JH, Kleindienst TE, Lin YH, Rubitschun CL, Surratt JD, Hayes PL, Jimenez JL, Goldstein AH. Detailed chemical characterization of unresolved complex mixtures in atmospheric organics: insights into emission sources, atmospheric processing, and secondary organic aerosol formation. J Geophys Res-Atmos. 2013;118(12):6783–6796. [Google Scholar]
- Cleveland MJ, Ziemba LD, Griffin RJ, Dibb JE, Anderson CH, Lefer B, Rappenglueck B. Characterization of urban aerosol using aerosol mass spectrometry and proton nuclear magnetic resonances. Atmos Environ. 2012;54:511–518. [Google Scholar]
- Collister JW, Rieley G, Stern B, Eglinton G, Fry B. Compound-specific delta-C-13 analyses of leaf lipids from plants with differing carbon-dioxide metabolisms. Org Geochem. 1994;21:619–627. [Google Scholar]
- Cristofanelli P, Fierli F, Marinoni A, Calzolari E, Duchi R, Burkhart J, Stohl A, Maione M, Arduini J, Bonasoni P. Influence of biomass burning and anthropogenic emissions on ozone, carbon monoxide and black carbon at the Mt. Cimone GAW-WMO global station (Italy, 2165 m.a.s.l.) Atmos Chem Phys. 2013;13:15–30. [Google Scholar]
- Decesari S, Facchini MC, Matta E, Lettini F, Mircea M, Fuzzi S, Tagliavini E, Putaud JP. Chemical features and seasonal variation of fine aerosol water-soluble organic compounds in the Po Valley. Italy Atmos Environ. 2001;35:3691–3699. [Google Scholar]
- Decesari S, Facchini MC, Fuzzi S, McFiggans GB, Coe H, Bower KN. The water-soluble organic component of size-segregated aerosol, cloud water and wet depositions from Jeju Island during ACE-Asia. Atmos Environ. 2005;39:211–222. [Google Scholar]
- Decesari S, Fuzzi S, Facchini MC, Mircea M, Emblico L, Cavalli F, Maenhaut W, Chi X, Schkolnik G, Falkovich A, Rudich Y, Claeys M, Pashynska V, Vas G, Kourtchev I, Vermeylen R, Hoffer A, Andreae MO, Tagliavini E, Moretti F, Artaxo P. Characterization of the organic composition of aerosols from Rondonia, Brazil, during the LBA-SMOCC 2002 experiment and its representation through model compounds. Atmos Chem Phys. 2006;6:375–402. [Google Scholar]
- Decesari S, Mircea M, Cavalli F, Fuzzi S, Moretti F, Tagliavini E, Facchini MC. Source attribution of water-soluble organic aerosol by nuclear magnetic resonance spectroscopy. Environ Sci Technol. 2007;41:2479–2484. doi: 10.1021/es061711l. [DOI] [PubMed] [Google Scholar]
- Decesari S, Finessi E, Rinaldi M, Paglione M, Fuzzi S, Stephanou EG, Tziaras T, Spyros A, Ceburnis D, O’Dowd C, Dall’Osto M, Harrison RM, Allan J, Coe H, Facchini MC. Primary and secondary marine organic aerosols over the North Atlantic Ocean during the MAP experiment. J Geophys Res-Atmos. 2011;116:D22210. [Google Scholar]
- Ding X, Zheng M, Yu LP, Zhang XL, Weber RJ, Yan B, Russell AG, Edgerton ES, Wang XM. Spatial and seasonal trends in biogenic secondary organic aerosol tracers and water-soluble organic carbon in the southeastern United States. Environ Sci Technol. 2008;42(14):5171–5176. doi: 10.1021/es7032636. [DOI] [PubMed] [Google Scholar]
- DRI (Desert Research Institute), Standard Operating Procedure DRI Model. Thermal/Optical Carbon Analysis (OR/TOT) of Aerosol Filter Samples. 2001 Methods IMPROVE_A, DRI SOP #2–216r3 October 22 2012 (available online from: http://www3.epa.gov/ttnamti1/files/ambient/pm25/spec/DRI_SOPforIMPROVEAFINAL.pdf.
- Duarte RMBO, Santos EBH, Pio CA, Duarte AC. Comparison of structural features of water-soluble organic matter from atmospheric aerosols with those of aquatic humic substances. Atmos Environ. 2007;41(37):8100–8113. [Google Scholar]
- EPA Positive Matrix Factorization (PMF) 3.0 Fundamentals and User Guide. US Environmental Protection Agency, Office of Research and Development; Washington, DC: EPA 600/R-17/108 2014. [Google Scholar]
- Ernvens B, Feingold G, Kreidenweis SM. Influence of water-soluble organic carbon on cloud drop number concentration. J Geophys Res Atmos. 2005;110(D18) http://dx.doi.org/10.1029/2004JD005634Art. No. D18211. [Google Scholar]
- Fuzzi S, Andreae MO, Huebert BJ, Kulmala M, Bond TC, Boy M, Doherty SJ, Guenther A, Kanakidou M, Kawamura K, Kerminen V, Lohmann U, Russell LM, Poeschl U. Critical assessment of the current state of scientific knowledge, terminology, and research needs concerning the role of organic aerosols in the atmosphere, climate, and global change. Atmos Chem Phys. 2006;6:2017–2038. [Google Scholar]
- Graham B, Mayol-Bracero OL, Guyon P, Roberts GC, Decesari S, Facchini MC, Artaxo P, Maenhaut W, Koll P, Andreae MO. Water-soluble organic compounds in biomass burning aerosols over Amazonia - 1. Characterization by NMR and GC-MS. J Geophys Res Atmos. 2002;107:8047. [Google Scholar]
- Hand JL, Schichtel BA, Pitchford M, Malm WC, Frank NH. Seasonal composition of remote and urban fine particulate matter in the United States. J Geophys Res Atmos. 2012:117. http://dx.doi.org/10.1029/2011JD017122Art. No. D05209.
- Heo JB, Dulger M, Olson MR, McGinnis JE, Shelton BR, Matsunaga A, Sioutas C, Schauer JJ. Source apportionments of PM2.5 organic carbon using molecular marker positive matrix factorization and comparison of results from different receptor models. Atmos Environ. 2013;73:51–61. [Google Scholar]
- Ho KF, Lee SC, Cao JJ, Li YS, Chow JC, Watson JG, Fung K. Variability of organic and elemental carbon, water soluble organic carbon, and isotopes in Hong Kong. Atmos Chem Phys. 2006;6:4569–4576. [Google Scholar]
- Lee JH, Hopke PK. Apportioning sources of PM2.5 in St. Louis, MO using speciation trends network data. Atmos Environ. 2006;40:360–377. [Google Scholar]
- Lin G, Silman S, Penner JE, Ito A. Global modeling of SOA; the use of different mechanism for aqueous-phase formation. Atmos Chem Phys. 2014;14(11):5451–5475. [Google Scholar]
- Mircea M, Facchini MC, Decesari S, Fuzzi S, Charlson RJ. The influence of the organic aerosol component on CCN supersaturation spectra for different aerosol types. Tellus. 2002;54(1):74–81. [Google Scholar]
- Nelson ST. A simple, practical methodology for routine VSMOW/SLAP normalization of water samples analyzed by continuous flow methods. Rapid Commun Mass Sp. 2000;14:1044–1046. doi: 10.1002/1097-0231(20000630)14:12<1044::AID-RCM987>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Paatero J, Hatakka J, Ikäheimonen TK, Klemola S, Salminen S, Lehto J. Radiological protection in transition. Swedish Radiation Protection Authority; Stockholm: 2005. Analysis intercomparison of lead-210 in aerosol filters; p. 55.p. 15. SSI Rapport. [Google Scholar]
- Paatero P, Tapper U. Positive matrix factorization: a non-negative factor model with optimal utilization of error estimates of data values. Environmetrics. 1994;5(2):111–126. [Google Scholar]
- Paatero P, Hopke PK, Song XH, Ramadan Z. Understanding and controlling rotation in factor analytic models. Chemom Intell Lab Syst. 2002;60:253–264. [Google Scholar]
- Paglione M, Kiendler-Scharr A, Mensah AA, Finessi E, Giulianelli L, Sandrini S, Facchini MC, Fuzzi S, Schlag P, Piazzalunga A, Tagliavini E, Henzing JS, Decesari S. Identification of humic-like substances (HULIS) in oxygenated organic aerosols using NMR and AMS factor analyses and liquid chromatographic techniques. Atmos Chem Phys. 2014;14(1):25–45. [Google Scholar]
- Pal KA, Watson C, Pirela SV, Singh D, Chalbot MCG, Kavouras I, Demokritou P. Linking exposures of particles released from nano-enabled products to toxicology: an integrated methodology for particle sampling, extraction, dispersion and dosing. Tox Sci. 2015;146(2):321–333. doi: 10.1093/toxsci/kfv095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, Jacod DL, Chin M, Martin RV. Sources of carbonaceous aerosols over the United States and implications for natural visibility. J Geophys Res Atmos. 2003:108. http://dx.doi.org/10.1029/2002JD003190Art. No. 4355.
- Ruehl CR, Wilson KR. Surface organic monolayers control the hygroscopic growth of submicrometer particles at high relative humidity. J Phys Chem A. 2014;118(22):3952–3966. doi: 10.1021/jp502844g. [DOI] [PubMed] [Google Scholar]
- Shakya KM, Place PF, Jr, Griffin RJ, Talbot RW. Carbonaceous content and water-soluble organic functionality of atmospheric aerosols at a semi-rural New England location. J Geophys Res Atmos. 2012;117:D03301. [Google Scholar]
- Shen X, Lee T, Guo J, Wang X, Li P, Xu P, Wang Y, Ren Y, Wang W, Wang T, Li Y, Carn SA, Collett JL., Jr Aqueous phase sulfate production in clouds in eastern China. Atmos Environ. 2012;62:502–511. [Google Scholar]
- Song XH, Polissar AV, Hopke PK. Sources of fine particle composition in the northeastern US. Atmos Environ. 2001;2001(5):5277. [Google Scholar]
- Stone EA, Snyder DC, Sheesley RJ, Sullivan AP, Weber RJ, Schauer JJ. Source apportionment of fine organic aerosol in Mexico City during the MILAGRO experiment 2006. Atmos Chem Phys. 2008;8(5):1249–1259. [Google Scholar]
- Turpin BJ, Lim HJ. Species contributions to PM2.5 mass concentrations: revisiting common assumptions for estimating organic mass. Aer Sci Technol. 2001;35:602–610. [Google Scholar]
- Vargas V, Chalbot MC, O’Brien R, Nikolich G, DuBois D, Etyemezian V, Kavouras IG. The impact of anthropogenic volatile organic compounds sources on ozone in an urban area. Environ Chem. 2014;11(4):445–458. [Google Scholar]
- Villalobos AN, Barraza F, Jorquera H, Schauer JJ. Chemical speciation and source apportionment of fine particulate matter in Santiago, Chile, 2013. Sci Total Environ. 2013;512–513:133–142. doi: 10.1016/j.scitotenv.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Widory D, Roy S, Le Moullec Y, Goupil G, Cocherie A, Geuerrot C. The origins of atmospheric particles in Paris: a view through carbon and lead isotopes. Atmos Environ. 2004;38:953–961. [Google Scholar]
- Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia J, Jia L, Cruz JA, Lim E, Sobsey CA, Shrivastava S, Huang P, Liu P, Fang L, Peng J, Fradette R, Cheng D, Tzur D, Clements M, Lewis A, De Souza A, Zuniga A, Dawe M, Xiong Y, Clive D, Greiner R, Nazyrova A, Shaykhutdinov R, Li L, Vogel HJ, Forsythe I. HMDB: a knowledgebase for the human metabolome. Nucleic Acid Res. 2009;37:D603–D610. doi: 10.1093/nar/gkn810. http://dx.doi.org/10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.