

SUMMARY
During valvulogenesis, globular endocardial cushions elongate and remodel into highly organized thin fibrous leaflets. Proper regulation of this dynamic process is essential to maintain unidirectional blood flow as the embryonic heart matures. In this study, we tested how mechanosensitive small GTPases, RhoA and Rac1, coordinate atrioventricular valve (AV) differentiation and morphogenesis. RhoA activity and its regulated GTPase-activating protein FilGAP are elevated during early cushion formation but decreased considerably during valve remodeling. In contrast, Rac1 activity was nearly absent in the early cushions but increased substantially as the valve matured. Using gain- and loss-of-function assays, we determined that the RhoA pathway was essential for the contractile myofibroblastic phenotype present in early cushion formation but was surprisingly insufficient to drive matrix compaction during valve maturation. The Rac1 pathway was necessary to induce matrix compaction in vitro through increased cell adhesion, elongation, and stress fiber alignment. Facilitating this process, we found that acute cyclic stretch was a potent activator of RhoA and subsequently downregulated Rac1 activity via FilGAP. On the other hand, chronic cyclic stretch reduced active RhoA and downstream FilGAP, which enabled Rac1 activation. Finally, we used partial atrial ligation experiments to confirm in vivo that altered cyclic mechanical loading augmented or restricted cushion elongation and thinning, directly through potentiation of active Rac1 and active RhoA, respectively. Together, these results demonstrate that cyclic mechanical signaling coordinates the RhoA to Rac1 signaling transition essential for proper embryonic mitral valve remodeling.
Graphical Abstract
INTRODUCTION
Many valve-related disorders originate during embryonic development. Although failure to initiate the formation of valves is uniformly lethal in early gestation, clinically serious malformations arise from improper structural maturation of the valvuloseptal apparatus and outflow tract [1]. These can be immediately life threatening at birth or more subtly impair the long-term durability and homeostatic remodeling capacity of the valve [2]. Although the regulatory events initiating endocardial cushion formation are well known, mechanistic understanding of the clinically important later phases of cushion remodeling and leaflet thinning is limited. Cushion compaction, elongation, and deposition of fibrillar collagen networks are critically important to maintain biomechanical competency under increasing cardiac loads [3–5]. Several genetic deletions associate with poorly condensed, non-elongated mitral valves and persistence of immature cushion cell phenotypes in vivo [6, 7]. However, the delineation of their functional roles independent of or in concert with the continuous mechanical stimulation remains challenging. Identification of mechanobiological mechanisms during embryonic valve remodeling is therefore crucial to advance new strategies to correct defective valve remodeling.
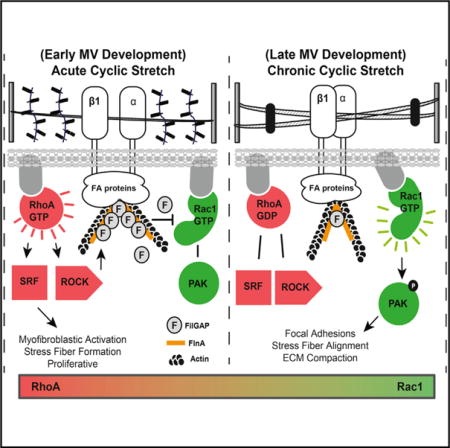
Cells sense their external mechanical environment through basal adhesion proteins (e.g., cadherins and integrins), apical surface components (e.g., stretch-activated channels), and cytoskeletal filaments, which can respond to both acute and chronic stimuli [8, 9]. A commonly utilized mechanical signal transduction involves activation of the Rho family of small GTPase proteins, specifically RhoA and Rac1. Mechanical insults cause these membrane-bound G proteins to become active through binding GTP, which then mediate rapid cytoskeletal rearrangements and/or downstream transcriptional activity. RhoA and Rac1 can act in opposing and complementary manners to control cell migration, differentiation, and proliferation, with the net responses dependent on the spatial and temporal dynamics of GTPaseactivity [10–12]. Almost all of our mechanistic understanding of GTPase coordination has been studied using 2D-cultured cell lines. Little is known how these behaviors orchestrate cell differentiation and tissue remodeling in 3D culture or in vivo. Rho kinase inhibition has been found to impair endocardial cushion mesenchyme migration, differentiation, and response to flow in vitro [13–15], but whether and how RhoA and Rac1 activity are coordinated by mechanical signaling to control valve remodeling is unknown. In this study, we recognized the distinct expression patterns and the functional roles of both RhoA and Rac1 during embryonic valve maturation. Importantly, we identified a new mechanobiological program by which the duration of cyclic stretch transitions between the activation of RhoA (acute) to Rac1 (chronic) through regulation of FilGAP in vitro. We further confirmed that cyclic loading coordinates valvular remodeling through regulation of RhoA and Rac1 activity in vivo.
RESULTS
Active RhoA and Rac1 Patterns with AV Progenitor Cell Differentiation and Matrix Remodeling
Native profiles of total and active (GTP-bound) Rac1 and RhoA in the developing left atrioventricular valve (AV) (HH25, HH36, and HH40) were evaluated using ELISA and immunofluorescence (whole mount) on freshly isolated tissue. We assessed the myofibroblastic phenotype of AV progenitor cells at each stage using markers for alpha-smooth muscle actin (aSMA—ACTA2 gene product) and serum response factor (SRF). ACTA2 is incorporated into contractile filaments prominently involved in myofibroblastic differentiation during valve remodeling and wound contraction [16–18]. SRF is a transcriptional regulator of ACTA2, and nuclear localization of SRF directly correlates with ACTA2 expression [19]. We determined that ACTA2, SRF, and active RhoA are all robustly expressed in the AV cushions at HH25 but decreased substantially during valve maturation (Figures 1A, 1B, and 1D). The expression of the mesenchymal intermediate filament vimentin (VIM) remained unchanged (Figure 1B, inset). Conversely, active Rac1 was low in the HH25 AV cushion but significantly increased during development (Figures 1C and 1D). Phospho-PAK1 (pPAK1—a downstream effector of activated Rac1 signaling) [20] and β1 integrin levels were elevated exclusively in later-stage AV remodeling (Figure S1B). Collectively, these results support that active RhoA is substantial during early AV cushion formation but abates in favor of active Rac1 as the cushion matures into thin leaflets. The early myofibroblast phenotype correlates with RhoA-GTP and ACTA2, whereas the late quiescent fibroblastic phenotype associates with Rac1-GTP (Figure 1E).
Figure 1. Native Profiles in the Developing Left AV Reveal a Divergence of RhoA and Rac1 Pathways in HH25, HH36, and HH40 Avian Embryos.
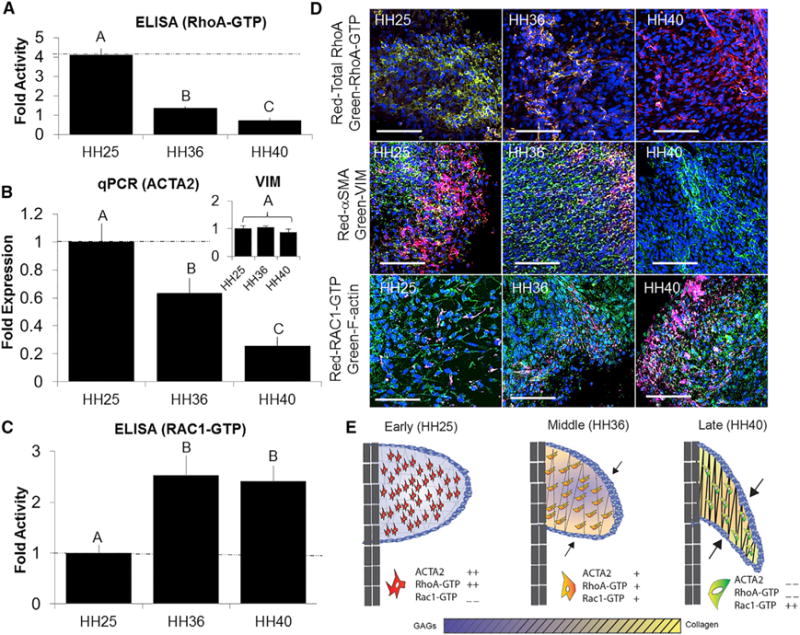
(A) Active RhoA was quantified using ELISA, and samples were normalized against total RhoA.
(B) mRNA expression of ACTA2 and VIM (inset) was quantified using real-time PCR.
(C) Active Rac1 was quantified using ELISA, and samples were normalized against total DNA.
(D) Whole-mount immunofluorescence of RhoA/RhoA-GTP (red/green, top row), ACTA2/VIM (red/green, middle row), and Rac1-GTP/F-actin (red/green, bottom row).
(E) Summary schematic combining the evolution of atrioventricular cushion morphology, ECM composition, and cell phenotype markers.
All samples were counterstained for nuclei using Draq5 (blue). Scale bars represent 100 μm (D). Error bars show ± SEM; n ≥ 6. One-way ANOVA with Tukey’s post hoc was performed. Different letters indicate statistical significance p < 0.05 from each other. See also Figure S1.
Matrix Compaction Potential of AV Progenitor Cells Is Cell Autonomous and Stage Dependent
We determined that the native thickness of the left AV primordia decreased considerably from HH25 to HH40 (Figure S1D). We hypothesized that leaflet thickness was a function of the matrix compaction potential in AV progenitor cells, which in turn could be stage dependent. We then freshly isolated resident cells from left AV tissue at HH25, HH36, and HH40 and disbursed them within 3D collagen hydrogels. After 24 hr, constructs were released and compaction area was measured over the next 5 days. We found that cell-mediated matrix compaction was stage dependent and significantly increased with more-mature cells (Figures S1C–S1E). Given that earlier-stage cushion mesenchymes are more proliferative than at later stages [13, 18, 21], these findings support that AV leaflet compaction is not associated with proliferation but rather is controlled by stage-specific autonomous behavior.
RhoA Activity Is Induced in AV Progenitor Cells by Mechanical Stress but Is Mitigated by Chronic Cyclic Stretch
Previous work has shown that mechanical force can regulate RhoA in AV cushion mesenchyme [10, 15]. This led us to initially hypothesize that cyclic stretch governs resident cell differentiation by promoting a myofibroblast phenotype via RhoA. We disbursed freshly isolated HH40 AV progenitor cells within collagen hydrogels and were subsequently stretched at 20% area strain at 2.1 Hz for up to 48 hr (Figure 2A). Mechanically anchored (CTRL-MA) gels were employed as non-stretched controls that develop mechanical stress, and free-floating (CTRL-FF) gels were employed as mechanically stress-free control environmental conditions [22–24]. ACTA2 and SRF mRNA expression increased with cyclic stretch (STIM) at 12 hr but surprisingly was downregulated over time. From 24 hr onward, neither gene was expressed differently from cells cultured within the stress-free (CTRL-FF) gels (Figures 2B and 2C).
Figure 2. Chronic Mechanical Stretch Mitigates Myofibroblastic Activation in HH40 AV Progenitor Cells.
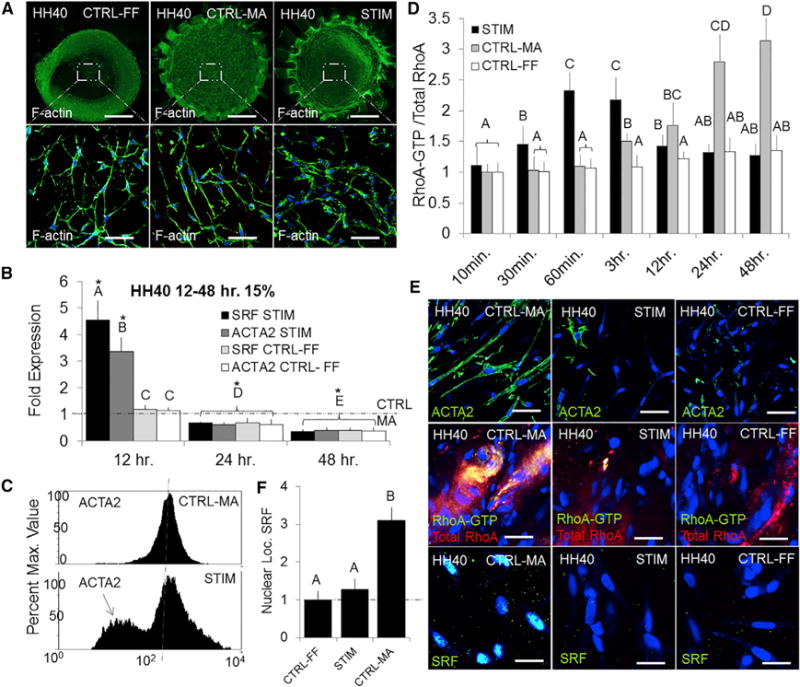
(A) Cells were isolated from left AV tissue and seeded into our previously described bioreactor. Free-floating (CTRL-FF liberated after 24 hr) and mechanically anchored (CTRL-MA confined compaction) gels were used as control conditions. Stretched gels (STIM) were allowed to compact for 24 hr and stimulated at 20% area strain at 2.1 Hz (average frequency during development).
(B) mRNA expression for SRF and ACTA2 was measured after 12, 24, and 48 hr of stretch using real-time PCR.
(C) Flow cytometry quantified ACTA2 protein expression in CTRL-MA and STIM conditions (10,000 cells).
(D) Dynamic active RhoA was quantified using ELISA for CTRL-MA, CTRL-FF, and STIM conditions. Data were normalized against total RhoA and then the baseline CTRL-MA (0 min) condition.
(E) Immunofluorescence for ACTA2 (green, top row), RhoA-GTP and RhoA (green and red, middle row), and SRF (green, bottom row) were conducted on fixed collagen constructs at 48 hr in CTRL-MA, CTRL-FF, and STIM conditions.
(F) Nuclear localization of SRF was quantified using ImageJ.
Scale bars represent 500 μm (A) and 20 μm (E). Error bars show ± SEM; n ≥ 4. One-way ANOVA with Tukey’s post hoc was performed. Different letters indicate statistical significance p < 0.05 from each other, and asterisks indicate significance from control. See also Figure S2.
We next performed a time course study using ELISA to quantify active RhoA dynamics in these different mechanical stress culture environments. Normalization for each sample was directly compared against total RhoA and then normalized to the initial (0 min) stressed (CTRL-MA) condition. Active RhoA levels during stretch (STIM) conditions rapidly increased with cyclic loading (peaking within 60 min) and then decreased to stress-free (CTRL-FF) levels thereafter. Conversely, stressed (CTRL-MA) gels increased active RhoA levels at later time points (Figure 2D). Reduced ACTA2 expression, SRF nuclear localization, and active RhoA levels at 48 hr were confirmed via immunofluorescence (Figure 2E). To quantify the levels of mechanical tension in stressed (CTRL-MA) gels, we developed a 3D culture system using a micro-spring as a cantilever force transducer. Tension increased in stressed (CTRL-MA) gels during culture time and strongly correlated with active RhoA (Figure S2). Together, these results establish that AV cushion cell-mediated 3D matrix tension activates RhoA and transitions toward a myofibroblastic phenotype. During cyclic stretch, RhoA activity is time dependent such that chronic loading reverts to baseline levels and inhibits the myofibroblastic differentiation.
Active RhoA Is Necessary and Sufficient for Myofibroblastic Differentiation of AV Progenitor Cells
We next performed gain- and loss-of-function experiments to determine whether active RhoA was required for myofibroblastic activation. As shown earlier, AV cushion cells in stress-free (CTRL-FF) gels express low levels of active RhoA, whereas stressed (CTRL-MA) gels express high levels of active RhoA after 48 hr. We therefore employed these conditions as negative and positive controls for RhoA activity, respectively. Transfection of HH40 AV progenitor cells with constitutively active RhoA (CA-RhoA) increased ACTA2, SRF, and CyclinB2 mRNA expression statistically significant from stress-free (CTRL-FF) gels, but not to the degree of stressed (CTRL-MA) gels. Conversely, dominant-negative RhoA (DN-RhoA)-transfected AV cushion mesenchyme cultured in stressed (CTRL-MA) gels significantly reduced ACTA2, SRF, and CyclinB2 mRNA expression comparable to stress-free (CTRL-FF) gels alone (Figures 3A and 3B). These responses were also confirmed at the protein level via immunofluorescence in stress-free (CTRL-FF) and stressed (CTRL-MA) conditions (Figures 3C and 3D). Comparing myofibroblastic differentiation marker expression directly against active RhoA (GTP-bound) for these conditions indicated that CTRL-MA conditions enhanced myofibroblastic activation above that explained by RhoA alone (Figure S3). These results suggest that other feedback mechanisms downstream of RhoA may also act to increase myofibroblastic differentiation, for example, adhesion or soluble signaling [25]. Hence, the complexity of our biological system is important to note. Next, cells were transfected with CA-RhoA and cultured during static stressed (CTRL-MA) or cyclic stretch (STIM) conditions for 48 hr. CA-RhoA overwhelmed the inhibition of myofibroblastic differentiation by chronic cyclic stretch to maintain myofibroblastic phenotype similar to that of stressed (CTRL-MA) gels (Figures 3E and 3F). Together, these results establish that active RhoA is essential for myofibroblastic differentiation and chronic cyclic stretch acts upstream to inhibit this process via reduction of active RhoA.
Figure 3. Myofibroblastic Activation in HH40 AV Progenitor Cells Is Dependent on Active RhoA.
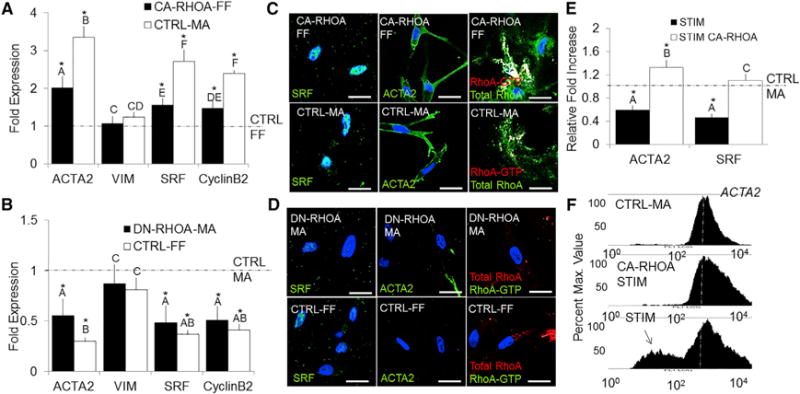
(A and B) HH40 cells were isolated from left AV tissue and immediately transfected with CA-RhoA or DN-RhoA (Neon Transfection system). mRNA expression for ACTA2, VIM, SRF, and CyclinB2 was quantified via real-time PCR and compared to CTRL-MA or CTRL-FF conditions.
(C and D) Immunofluorescence of SRF (green, left column), ACTA2 (green, middle column), and RhoA-GTP/RhoA (green/red, right column) in the collagen gels for each conditions.
(E) Transfected CA-RhoA cells were stretched (STIM) for 48 hr at 2.1 Hz, and mRNA expression of ACTA2 and SRF was quantified using real-time PCR.
(F) Flow cytometry was used to quantify ACTA2 protein expression in CTRL-MA, STIM, and CA-RhoA-STIM conditions (10,000 cells).
All samples were counterstained for nuclei using Draq5 (blue). Scale bars represent 10 μm (C and D). Error bars show ± SEM; n ≥ 4. One-way ANOVA with Tukey’s post hoc was performed. Different letters indicate statistical significance p < 0.05 from each other, and asterisks indicate significance from control. See also Figure S3.
Active Rac1 Promotes Matrix Compaction through Increased Adhesion, Focal Adhesion Formation, and Stress Fiber Alignment
We next tested whether RhoA and/or Rac1 mediated matrix compaction. RhoA activity (via CA-RhoA or DN-RhoA transfection, respectively) had no effect on the compaction potential as compared to controls (Figure 4A). CA-Rac1, however, significantly enhanced compaction, whereas DN-Rac1 inhibited compaction. Interestingly, the combination of CA-Rac1 with DN-RhoA inhibited compaction similar to controls (Figure 4B). Furthermore, ROCK inhibition was capable of inhibiting compaction by AV progenitor cells proportional to their stage-specific activity (Figure S4A). These results emphasize that basal levels of RhoA activity are required to sustain Rac1-induced matrix compaction. We next investigated whether Rac1 affected cell adhesion and/or stress fiber generation, which may contribute to the underlying tissue compaction and remodeling [26]. CA-Rac1 transfection increased the percentage of adherent AV mesenchyme cells in a time-dependent manner compared to controls, whereas DN-Rac1 transfection reduced adhesion (Figure 4C). Additionally, transfection with CA-Rac1 increased the number of focal adhesions at the leading edge of lamellipodium, which was determined by co-localization of vinculin and F-actin along the periphery. Moreover, F-actin stress fibers were highly aligned to the long axis of the cell, indicating elongation of the cell (Figure 4D). DN-Rac1 had the opposite effect: reduced adhesion and focal adhesion formation; randomly aligned actin filaments; and no discernable cell orientation (Figure 4E). Concurrently, CA-Rac1-transfected HH40 AV progenitor cells increased β1 integrin mRNA expression regardless of culture conditions (CTRL-MA or CTRL-FF), whereas DN-Rac1 had the opposite effect (Figures S4B–S4D). These results establish that active Rac1 increases integrin expression and focal adhesion formation, leading to enhanced cell adhesion and cell alignment that accompanies traction force generation.
Figure 4. Matrix Compaction Potential of HH40 AV Progenitor Cells Is Dependent on Rac1 Activity.
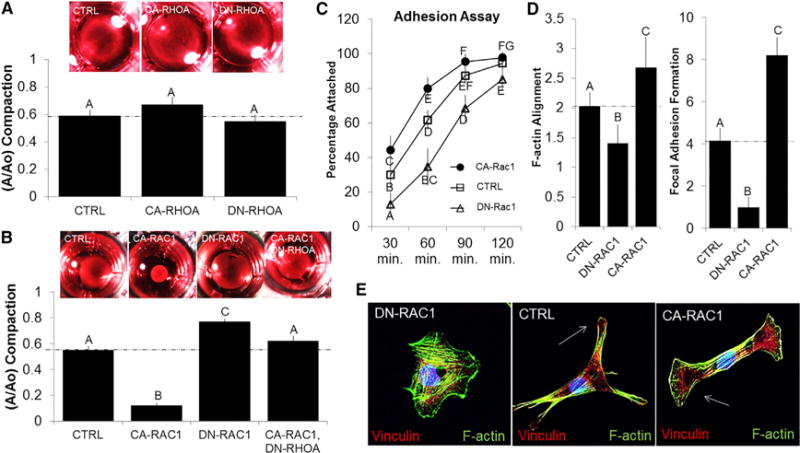
(A and B) HH40 cells were isolated from left AV tissue and immediately transfected with CA-RhoA, DN-RhoA, CA-Rac1, DN-Rac1, or CA-Rac1+DN-RhoA (Neon Transfection system). Cells were then embedded into collagen hydrogels and allowed to compact for 24 hr. Hydrogel compaction was measured by current area over initial area (A/Ao) and determined at the end of 48 hr. (A) RhoA activity was insufficient to drive compaction, whereas (B) Rac1 activity was capable but requires some basal level of RhoA activity to be present.
(C) An adhesion test was performed, and the percentage of adherent cells was quantified at each time interval after application of the orbital shaker.
(D) After 24 hr, the extent of F-actin alignment and focal adhesions were quantified for each condition.
(E) Focal adhesions and F-actin alignment were visualized using immunofluorescence (note the distribution angle of stress fibers alignment within CA-Rac1).
All samples were counterstained for nuclei using Draq5 (blue). Error bars show ± SEM; n ≥ 4. One-way ANOVA with Tukey’s post hoc was performed. Different letters indicate statistical significance p < 0.05. See also Figure S4.
Active Rac1 Is Elevated during Chronic Cyclic Stretch and Is Dependent on FilGAP
FilGAP is a GTPase-activating protein specific for Rac that is activated by RhoA/ROCK at sites of cell adhesions [27]. Previous studies have shown that quasi-static stretch directly modulates FilGAP in minimalist filament network models and in cell lines via its targeting by FlnA [28, 29]. We first profiled FilGAP/FlnA levels in developing AVs and found that FilGAP was distributed extensively in (HH25) the cushions but decreased as the leaflet compacted and matured (HH36 and HH40; Figure 5A). FilGAP largely co-localized with FlnA at all stages of development (Figure S5B). We therefore hypothesized that mechanical stress modulates active RhoA/Rac1 via FilGAP in a time-dependent manner. Using HH40 AV progenitor cells, FilGAP was reduced in stress-free (CTRL-FF) gels, at both acute (60 min) and chronic (24 hr) time points. Conversely, FilGAP was elevated in stressed (CTRL-MA) gels. During cyclic stretch, FilGAP was elevated at the acute time point (consistent with active RhoA and inactive Rac1), whereas the opposite occurred at the chronic time point (consistent with inactive RhoA and active Rac1; Figure 5B). To readily identify the trend in FilGAP levels, we quantified mean intensity per cell in the immunofluorescence images using ImageJ (Figure S5C).
Figure 5. Active Rac1 Increases during Chronic Cyclic Stretch and Is Dependent on FilGAP.
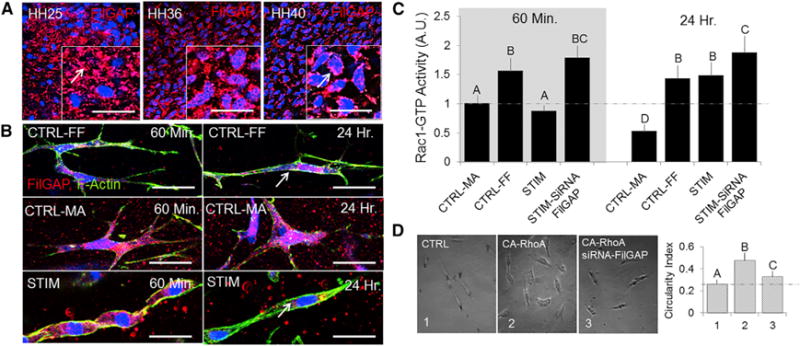
(A) Native whole-mount tissue from HH25, HH36, and HH40 AV tissue was immunostained for FilGAP (red).
(B) HH40 AV progenitor cells were subjected to cyclic stretch for either acute (60 min) or chronic (24 hr) time points, and gel constructs were immunostained for FilGAP (red, FilGAP; green, actin).
(C) Active Rac1 levels were quantified using ELISA during cyclic stretch and in the presence of siRNA FilGAP for either acute (60 min) or chronic (24 hr) time points.
(D) Cell morphology was analyzed by tracing cell shape and quantified using a circularity index.
All fluorescent samples were counterstained for nuclei using Draq5 (blue). Scale bars represent 20 μm (A and B). Error bars show ± SEM; n ≥ 4. One-way ANOVA with Tukey’s post hoc was performed. Different letters indicate statistical significance p < 0.05. See also Figure S5.
We next assessed Rac1 activity during these treatment conditions. Stressed (CTRL-MA) gels with DN-RhoA and siRNA FilGAP significantly increased active Rac1 at 24 hr. In stress-free (CTRL-FF) gels, CA-RhoA transfection deactivated Rac1 as expected, but CA-RhoA combined with siRNA FilGAP also significantly elevated active Rac1 (Figure S5A). Active Rac1 was unchanged with acute stretch (60 min), whereas chronic cyclic stretch (24 hr) elevated active Rac1 levels significantly. Chronic cyclic stretch combined with siRNA FilGAP treatment synergistically elevated Rac1 activity (Figure 5C). Consistent with these results, transfection with CA-RhoA increased the circularity of resident cells, thus decreasing cellular alignment similar to that of HH25 mesenchyme [30]. Moreover, the elongated cell morphology (similar to HH40 AV progenitors) was restored with co-addition of siRNA FilGAP (Figure 5D). Together, these results demonstrate that FilGAP regulates Rac1 downstream of active RhoA and cyclic stretch mediates this process in a time-dependent manner.
Mechanical Perturbation In Vivo Disrupts the RhoA to Rac1 Transition during Valvular Remodeling
We performed partial atrial ligation experiments on ex-ovocultured chick embryos to determine the morphogenetic impact of altered cyclic loading on valvulogenesis in vivo [31, 32] (Figure 6A). RAL (right atrial ligation) treatment (increased left AV preload) significantly increased left AV area and left ventricular volume after 3 days as compared to sham controls (determined via micro-CT). Conversely, LAL (left atrial ligation)-treated embryos (decreased left AV preload) significantly decreased left AV area and left ventricular volume as compared to sham controls (Figures 6B and 6C). As anticipated from our aforementioned results, reduced hemodynamic load enhanced myofibroblastic activation in the left AV at 7 days (D7) (HH30). Expression of ACTA2 and SRF mRNA was significantly elevated in LAL compared to sham controls, whereas increased loading (via RAL) suppressed myofibroblastic differentiation in the left AV at D7 (Figure 6D). β1 integrin mRNA and protein expression increased with increased preload (RAL) and decreased with a reduction in preload (LAL). LAL-treated embryos increased active RhoA and FilGAP, whereas active Rac1 was decreased relative to sham controls (Figures 6D and 6E). Conversely, RAL-treated embryos decreased SRF nuclear localization, RhoA-GTP activity, and FilGAP levels, whereas active Rac1 was significantly elevated (Figure 6E). We further analyzed RAL-treated embryos for an additional 3 days (10 days total; LAL treatment results in significant lethality after D7 as previously reported) [32] to determine long-term effects on AV remodeling (Figure 6F). At both D7 and D10, ACTA2 expression and SRF nuclear localization significantly decreased and correlated with active RhoA in the left AV, whereas active Rac1 significantly increased (Figures 6G and 6H). Furthermore, the RAL-treated embryos at D7 and D10 increased in collagen versus GAG content ratio relative to sham controls (Figures S6A and S6B). Together, these results demonstrate that cyclic mechanical loading is critical for proper valve compaction and elongation in vivo. Decreased hemodynamic preload resulted in a significantly underdeveloped valve with persistent immature activated myofibroblast-like mesenchyme, which correlated with hyperactive RhoA and inhibition of Rac1. Conversely, increased preload accelerated valve remodeling and maturation concomitant with activated Rac1 and reduced FilGAP. These findings are consistent with our in vitro mechanistic experimentation and highlight the role of chronic cyclic stretch in mediating FilGAP to promote necessary Rac1-mediated fetal valve remodeling.
Figure 6. Hemodynamic Perturbation In Vivo Regulates the Divergence of RhoA and Rac1 Pathways during AV Development.
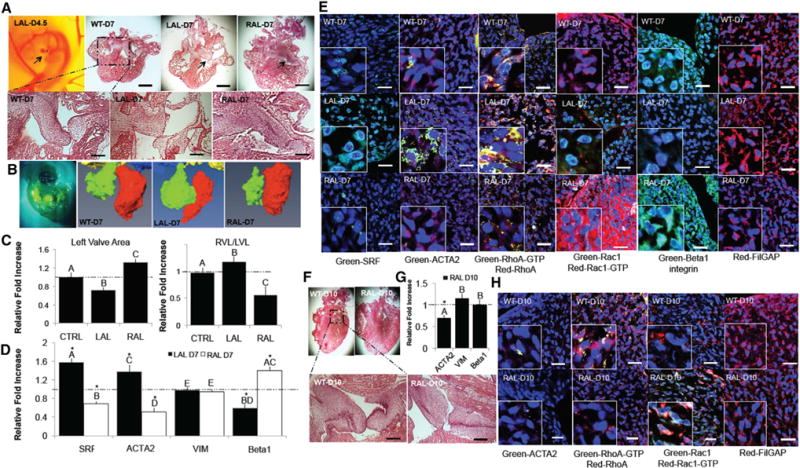
Left and right atriums of the heart were ligated using suture material between 4 and 5 days (LAL and RAL).
(A) H&E histology stains of the heart and left AV structures for the control and ligated conditions.
(B and C) Micro-CT was used to measure both the change in size of ventricular chambers but also left AV geometry (ultrasound also confirmed a significant decrease in time-averaged velocity and cardiac output) at D7.
(D) mRNA expression for SRF, ACTA2, VIM, and β1 integrin was quantified via real-time PCR at D7.
(E) Immunohistochemistry for SRF, ACTA2, RhoA-GTP, Rac1-GTP, β1 integrin, and FilGAP in resident valve cells at D7 of wild-type (top row), left atrial ligation (LAL; middle row), and right atrial ligation conditions (RAL; bottom row).
(F) RALs at D10 were analyzed. H&E histology was conducted on both the left AV and ventricular chamber structures.
(G) mRNA expression for ACTA2, VIM, and β1 integrin was quantified via real-time PCR at D10.
(H) Immunohistochemistry staining for ACTA2, RhoA-GTP, Rac1-GTP, and FilGAP in wild-type (top row), LAL (middle row), and RAL conditions (bottom row).
All samples were counterstained for nuclei using Draq5 (blue). Scale bars represent 750 μm (A) and 40 μm (E and H). Error bars show ± SEM; n ≥ 6. One-way ANOVA with Tukey’s post hoc w. Different letters indicate statistical significance p < 0.05 from each other, and asterisks indicate significance from control. See also Figure S6.
DISCUSSION
Proper tissue morphogenesis in development requires the acquisition of the correct tissue shape, cell phenotype, matrix architecture, and function. Deficiencies are rarely if ever seen in only one of these components, which supports that their evolution is interdependent. Elucidation of overarching governing mechanisms is therefore critical for informing possible strategies to correct defective morphogenesis. In this study, we clarify how mechanical signaling orchestrates endocardial cushion condensation into thin fibrous leaflets while simultaneously establishing fibroblastic quiescence. Using gain- and loss-of-function assays, we identify RhoA and Rac1 as key participants in mechanical regulation of embryonic valve remodeling. RhoA mediates myofibroblastic activation, and Rac1 is necessary for matrix compaction. More importantly, we found that chronic cyclic mechanical stretch in vitro inhibited myofibroblastic differentiation by deactivating RhoA and depleting the molecular switch Fil-GAP, which together enabled Rac1 activation and downstream signaling. Active Rac1 induced matrix compaction through augmenting focal adhesion formation, cell elongation, and stress fiber alignment (Figure 7). These effects were further confirmed in vivo using ligation experiments to regulate side-specific hemodynamic load. These results establish the importance of cyclic mechanical loads for directing valve progenitor differentiation and matrix compaction through coordination of RhoA/Rac1 signaling.
Figure 7. Summary of Phenotypic Changes Associated with Acute and Chronic Cyclic Stretch during Valve Maturation.
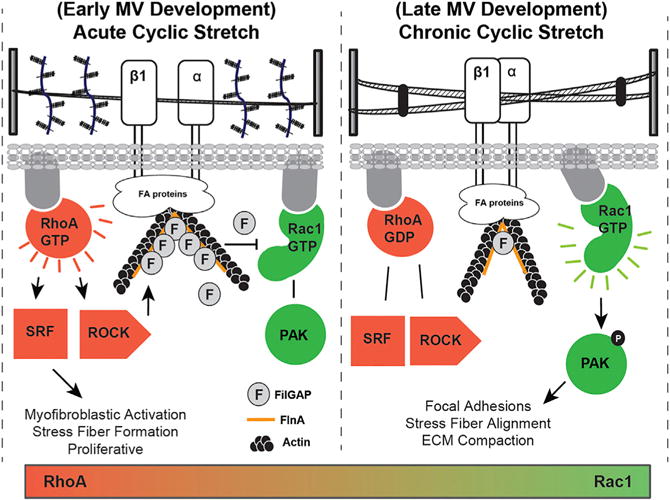
During acute cyclic stretch, resident valve cells activate the RhoA pathway and subsequent ROCK/FILGAP to deactivate Rac1. This leads to myofibroblastic activation, collagen production, and proliferation. During chronic cyclic stretch, FilGAP and active RhoA are reduced, which promotes active Rac1. Rac1 then initiates focal adhesion formation, elongation, and stress fiber alignment necessary for matrix compaction.
We determined the Rac1 pathway to be necessary for matrix compaction during valve maturation. This was initially surprising, as the RhoA pathway is a well-established driver of stress fiber formation and contractility [33, 34]. However, constitutively active Rac1 has been found to drive the formation of adhesions along the ventral surface of the cell body, which move toward those at the tip and result in contraction-like shortening and matrix compression at the base of lamellipodia [35]. Both the magnitude and direction of the adhesion motions correlate strongly with nearby matrix displacements, indicating the importance of cell polarity for force generation [36]. Additionally, fibroblast-specific deletion of Rac1 in mouse resulted in delayed cutaneous wound closure, with reduced collagen production and matrix compaction [26]. Consistent with these results, we determined that co-localization of actin fibers and vinculin along the cell periphery is dependent on Rac1 activity. Actin stress fibers were highly aligned within the cells only when Rac1 was active, indicating strong adhesion and contraction. The ability of Rac1 to drive matrix compaction explains in part the inverse relationship between matrix compaction and myofibroblastic differentiation during valve maturation. This is essential for developing a thin mature quiescent valve. Neither these studies nor our results here suggest that RhoA activity is completely dispensable for tissue remodeling to occur. Cyclic modulation of RhoA and Rac1 activity is common for long-term behaviors such as cardiac neural crest migration [37]. Rather, our results are consistent with cyclic stretch shifting the balance of RhoA/Rac1 signaling toward Rac1, which in turn promotes the valvular remodeling events we identified over time.
FilGAP and its close relatives are molecular switches that regulate the reciprocal inhibitory relationship between RhoA and Rac1 [38]. Rho/Rac signaling has been studied extensively in early embryonic patterning and differentiation events [37, 39], but their roles in clinically important later tissue-remodeling events are poorly understood. Our results confirm that FilGAP modulation of Rho/Rac signaling is required in progenitor mitral valve cells for proper valve compaction and elongation, and the regulatory mechanism was found to be dependent upon the duration of cyclic mechanical loading. Specifically, chronic cyclic stretch is a potent inhibitor of active RhoA and downstream FilGAP to enable Rac1-mediated signaling. Conversely, acute stretch enhances both active RhoA and FilGAP to inhibit the Rac1 pathway [40]. Speculating on these findings, initial loading most likely creates an acute spike in matrix tension to drive hyperactive levels of active RhoA and FilGAP similar to previous studies involving step changes in stretch [28, 29]. However, as the cyclic loading becomes chronic, cells adapt to and remodel the matrix environment to potentially relieve cytoskeletal tension and thereby progressively develop a quiescent phenotype [41]. Therefore, the chronic cyclic mechanical loading of the embryonic valves is an essential component to orchestrate valve sculpting and fibroblast quiescence.
Chronic cyclic stretch in a 3D environment is a unique mechanism to dissipate FilGAP, enabling Rac1 to dominate. Although the exact mechanism has yet to be established, we believe it could be through a number of possibilities. This includes dissociation from FlnA [28] and direct inactivation of FilGAP via ROCK (possible through phosphorylation sites) [27, 40], a chronic reduction in the overall amount of FilGAP (reduced mRNA expression), and/or upstream ubiquitin-proteasome machinery to restrict FlnA/Rho GTPase that is not yet identified [42, 43]. Furthermore, dynamics in the subcellular localization of FilGAP, FlnA, and GTPase components near adhesion sites/cytoskeletal junctions with stretch are most likely dependent on ECM remodeling in a time course manner [40]. Regardless of the specific process, the previously unrecognized role of time dependency of cyclic stretch is a powerful coordinator of GTPase signaling to drive 3D morphogenetic events. Therefore, control of this mechanobiological program may be a novel means to restore proper valve remodeling and maintain valve tissue homeostasis. Although mutations in FilGAP have not yet been identified in patients with congenital or acquired valve defects, mutations have been found in FlnA that disrupt GTPase regulation and weakening of FilGAP binding [44, 45].
In summary, these results establish that cyclic mechanical loading is essential for coordinating a transition from RhoA- to Rac1-based signaling via FilGAP to produce a thin organized valve leaflet with a quiescent phenotype. Unbalanced RhoA/Rac1 signaling via FilGAP may therefore be an important predictor of abnormal valve-remodeling capacity and susceptibility premature dysfunction. Additionally, altered hemodynamic signaling can possibly be used to control RhoA/Rac1 signaling to rescue tissue fate. These findings and innovative methodologies motivate and enable new mechanobiological hypotheses to link microenvironmental cues with genetic regulation for understanding tissue formation and remodeling.
EXPERIMENTAL PROCEDURES
Avian Valve Cell Progenitor Isolation and Culture
AV cushions (HH25) or remodeling left AV leaflets (HH36 or HH40) were dissected from the myocardium of embryonic chick hearts and digested via trypsin (0.025%; Invitrogen) to obtain pure populations of valve progenitor cells as previously described [46] and used directly in experiments (without passaging). Cells were pelleted via centrifugation and re-suspended within a neutralized collagen hydrogel (1.5 mg/ml) solution at a density of 400,000 cells/ml. Approximately 150 μl of gel was inoculated into culture wells, which solidified after 60 min. Cells were cultured in a base medium consisting of M199 (Invitrogen) supplemented with 5% chick serum (Hyclone), 250 μg/ml insulin, 25 μg/ml transferrin, and 250 μg/ml selenium (ITS; Sigma) and 1% penicillin/streptomycin as previously established for valve mesenchyme [13].
Static and Cyclic Stress Experiments
A subset of collagen gels were formed and cultured around rings of compression springs as previously described [22], creating a MA stressed condition [23]. Alternatively, gels were released from the springs after 24 hr of culture, creating a FF stress-free culture condition [16]. As a third condition, we applied cyclic stretch to 3D collagen hydrogels via a custom-designed bioreactor system as previously described [22]. Cell encapsulated gels solidified around compression springs over a 24 hr period, after which 20% cyclic equiaxial strain (area strain; 225 step size; 2 mm) was applied at 2.1 Hz, which approximated the in vivo HH25 embryonic valve cyclic strain profile (STIM) [47]. Experiments were conducted at both acute (<60 min) and chronic (up to 48 hr) time intervals. Molecular, cellular, and tissue level responses within each 3D mechanical environment were compared between CTRL-MA (mechanically anchored: stressed), CTRL-FF (free floating: unstressed), and STIM (cyclically stretched gels) conditions.
RhoA/Rac1 Activity and Gain-/Loss-of-Function Assays
Freshly isolated AV progenitors were electroporated in solution with plasmids encoding for constitutively active RhoA (CA-RhoA; Q63L), CA-Rac1 (Q61L), dominant-negative RhoA (DN-RhoA; T19N), or DN-Rac1 (T17N; all available through AddGene) using the Neon transfection system (Invitrogen) as previously described [48]. Cells were then embedded into collagen constructs and further cultured in 5% serum, antibiotic-free M199 for up to 48 hr after initial compaction. For ROCK inhibition, cultures were treated with Y-27632 (10 μM; Cell Signaling Technology) with media changed every 48 hr. RhoA and Rac1 activity in AV cushion mesenchyme was measured using ELISA activation assay kits (GLISA; Cytoskeleton) according to the manufacturer’s instructions. Briefly, cell extracts were added to 96-well plate coated with GST fusion of Rho-binding domain, which selectively binds GTP-bound (active) GTPases. After incubation with shaking at 4°C for 30 min, the plate was washed three times with washing buffer (Cytoskeleton), removing all GDP (inactive) RhoA GTPase. The substrate with captured GTP-bound RhoA was then incubated with specific anti-RhoA antibodies. The GTPase-antibody conjugates were detected with horseradish peroxidase (HRP)-conjugated secondary antibody and quantified using an Epoch Microplate Spectrophotometer (Biotek). Identical experiments were conducted to quantify active Rac1. Detection of RhoA-GTP or Rac1-GTP was analyzed in thin sections and 3D cultures using primary antibodies specific to GTP-bound RhoA or Rac1 (NewEast Biosciences) according to the manufacturer’s instructions. Antibody specificity was confirmed by direct comparison of GTP-specific RhoA or Rac1 immunofluorescence and ELISA quantification in AV progenitor cells using CA, wild-type, or DN transfection as well as dual labeling with antibodies specific to total or active RhoA and Rac1 GTPase (Figure S1A).
Cell Phenotype Assessment
Valve cell phenotype was assessed at both the gene and protein level (see the Supplemental Experimental Procedures for additional details). Briefly, gene expression was measured by extracting RNA using a QIAGEN total RNA purification kit (QIAGEN) and reverse transcribed to cDNA using the SuperScript III RT-PCR kit with oligo(dT) primer (Invitrogen). Samples were amplified using the SYBR Green PCR system (Bio-Rad) on a Bio-Rad CFX96 cycler. For visualization of proteins, collagen constructs were fixed in 4% PFA overnight at 4°C. Cells were incubated overnight with the appropriate primary/secondary antibodies, washed, and nuclear counterstained for subsequent confocal imaging. For quantification of protein expression, cells were trypsinized from the collagen constructs, fixed with 4% PFA for 10 min, and then preserved in 50% methanol/PBS. Cells were further washed and incubated with appropriate primary/secondary antibodies and scanned using a Coulter Epics XL-MCL Flow Cytometer (Coulter). Adhesion testing assay methods are included in the Supplemental Experimental Procedures.
In Vivo Atrial Ligation Experiments
Fertilized White Leghorn chicken eggs were incubated in a 38°C forced-draft incubator to Hamburger-Hamilton (HH) stage 21 (3.5 days; Hamburger and Hamilton). The embryo was cultured in an ex ovo platform previously described [49]. Briefly, an overhand knot of 10-0 nylon suture loop was placed across a portion of either the right or left atrium and tightened, partially constricting the left or right AV orifice, respectively (LAL and RAL). This diverted flow from the constricted inlet toward the untreated inlet, decreasing hemodynamic load on the one side and increasing it on the other [31, 32]. At D7 and D10, hearts and/or AVs were analyzed using RT-PCR, immunohistochemistry, and 3D micro-CT imaging.
Statistics
Results are expressed as mean ± SEM, with n ≥ 4 independent cultures per treatment condition and four to six dozen chick embryos pooled for each experiment. Statistical analysis was done using SAS and/or Excel software. Treatment effects were compared using either an ANOVA with Tukey post hoc paired tests or t test, and data were transformed when necessary to obtain equal sample variances. Differences between means were considered significant at p ≤ 0.05.
Supplementary Material
Highlights.
RhoA and Rac1 are key participants in embryonic valve morphogenesis
RhoA controls myofibroblastic activation during early valve development
Rac1 mediates matrix compaction through cell elongation and stress-fiber alignment
Mechanical stretch regulates the transition between RhoA and Rac1 via FilGAP
Acknowledgments
This research was supported in part by the NIH (HL110328 and HL118672 to J.T.B. and 1DP2OD004213 and 1R21EB016359 to S.K.), the American Heart Association (no. 0830384N to J.T.B.), the National Science Foundation (CBET-0955172 to J.T.B. and CMMI 1055965 to S.K.), the Leducq Foundation (to J.T.B. and R.N.), and the Marie Curie International Reintegration Grant within the 7th European Community Framework Programme, IRG276987 to H.C.Y.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, R.A.G. and J.T.B.; Methodology, R.A.G., H.C.Y., J.L.M., and J.T.B.; Investigation, R.A.G., H.Y.C., and J.T.B.; Validation, H.C.Y. and K.S.; Formal Analysis, R.A.G. and H.C.Y.; Writing – Original Draft, R.A.G. and J.T.B.; Writing – Review & Editing, R.A.G., S.K., R.N., and J.T.B.; Funding Acquisition, S.K., R.N., and J.T.B.; Resources, J.L.M. and S.K.; Supervision, R.N., S.K., and J.T.B.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.cub.2015.11.033.
References
- 1.Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–1048. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodwin RL, Nesbitt T, Price RL, Wells JC, Yost MJ, Potts JD. Three-dimensional model system of valvulogenesis. Dev Dyn. 2005;233:122–129. doi: 10.1002/dvdy.20326. [DOI] [PubMed] [Google Scholar]
- 4.Buskohl PR, Gould RA, Butcher JT. Quantification of embryonic atrioventricular valve biomechanics during morphogenesis. J Biomech. 2012;45:895–902. doi: 10.1016/j.jbiomech.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buskohl PR, Jenkins JT, Butcher JT. Computational simulation of hemodynamic-driven growth and remodeling of embryonic atrioventricular valves. Biomech Model Mechanobiol. 2012;11:1205–1217. doi: 10.1007/s10237-012-0424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakraborty S, Combs MD, Yutzey KE. Transcriptional regulation of heart valve progenitor cells. Pediatr Cardiol. 2010;31:414–421. doi: 10.1007/s00246-009-9616-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Vlaming A, Sauls K, Hajdu Z, Visconti RP, Mehesz AN, Levine RA, Slaugenhaupt SA, Hagège A, Chester AH, Markwald RR, et al. Atrioventricular valve development: new perspectives on an old theme. Differentiation. 2012;84:103–116. doi: 10.1016/j.diff.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475:316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ingber DE. Mechanical control of tissue morphogenesis during embryological development. Int J Dev Biol. 2006;50:255–266. doi: 10.1387/ijdb.052044di. [DOI] [PubMed] [Google Scholar]
- 10.Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- 11.Chauhan BK, Lou M, Zheng Y, Lang RA. Balanced Rac1 and RhoA activities regulate cell shape and drive invagination morphogenesis in epithelia. Proc Natl Acad Sci USA. 2011;108:18289–18294. doi: 10.1073/pnas.1108993108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rottner K, Hall A, Small JV. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr Biol. 1999;9:640–648. doi: 10.1016/s0960-9822(99)80286-3. [DOI] [PubMed] [Google Scholar]
- 13.Butcher JT, Norris RA, Hoffman S, Mjaatvedt CH, Markwald RR. Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev Biol. 2007;302:256–266. doi: 10.1016/j.ydbio.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tavares AL, Mercado-Pimentel ME, Runyan RB, Kitten GT. TGF beta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev Dyn. 2006;235:1589–1598. doi: 10.1002/dvdy.20771. [DOI] [PubMed] [Google Scholar]
- 15.Tan H, Biechler S, Junor L, Yost MJ, Dean D, Li J, Potts JD, Goodwin RL. Fluid flow forces and rhoA regulate fibrous development of the atrioventricular valves. Dev Biol. 2013;374:345–356. doi: 10.1016/j.ydbio.2012.11.023. [DOI] [PubMed] [Google Scholar]
- 16.Butcher JT, Barrett BC, Nerem RM. Equibiaxial strain stimulates fibroblastic phenotype shift in smooth muscle cells in an engineered tissue model of the aortic wall. Biomaterials. 2006;27:5252–5258. doi: 10.1016/j.biomaterials.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 17.Grinnell F, Ho CH. Transforming growth factor beta stimulates fibroblast-collagen matrix contraction by different mechanisms in mechanically loaded and unloaded matrices. Exp Cell Res. 2002;273:248–255. doi: 10.1006/excr.2001.5445. [DOI] [PubMed] [Google Scholar]
- 18.Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 19.Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem. 2001;276:341–347. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- 20.Revenu C, Athman R, Robine S, Louvard D. The coworkers of actin filaments: from cell structures to signals. Nat Rev Mol Cell Biol. 2004;5:635–646. doi: 10.1038/nrm1437. [DOI] [PubMed] [Google Scholar]
- 21.Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB. TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol. 1999;208:530–545. doi: 10.1006/dbio.1999.9211. [DOI] [PubMed] [Google Scholar]
- 22.Gould RA, Chin K, Santisakultarm TP, Dropkin A, Richards JM, Schaffer CB, Butcher JT. Cyclic strain anisotropy regulates valvular interstitial cell phenotype and tissue remodeling in three-dimensional culture. Acta Biomater. 2012;8:1710–1719. doi: 10.1016/j.actbio.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 2003;13:264–269. doi: 10.1016/s0962-8924(03)00057-6. [DOI] [PubMed] [Google Scholar]
- 24.Butcher JT, Nerem RM. Porcine aortic valve interstitial cells in three-dimensional culture: comparison of phenotype with aortic smooth muscle cells. J Heart Valve Dis. 2004;13:478–485. [PubMed] [Google Scholar]
- 25.Ren XD, Wang R, Li Q, Kahek LA, Kaibuchi K, Clark RA. Disruption of Rho signal transduction upon cell detachment. J Cell Sci. 2004;117:3511–3518. doi: 10.1242/jcs.01205. [DOI] [PubMed] [Google Scholar]
- 26.Liu S, Kapoor M, Leask A. Rac1 expression by fibroblasts is required for tissue repair in vivo. Am J Pathol. 2009;174:1847–1856. doi: 10.2353/ajpath.2009.080779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol. 2006;8:803–814. doi: 10.1038/ncb1437. [DOI] [PubMed] [Google Scholar]
- 28.Ehrlicher AJ, Nakamura F, Hartwig JH, Weitz DA, Stossel TP. Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature. 2011;478:260–263. doi: 10.1038/nature10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shifrin Y, Arora PD, Ohta Y, Calderwood DA, McCulloch CA. The role of FilGAP-filamin A interactions in mechanoprotection. Mol Biol Cell. 2009;20:1269–1279. doi: 10.1091/mbc.E08-08-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gould RA, Sinha R, Aziz H, Rouf R, Dietz HC, 3rd, Judge DP, Butcher J. Multi-scale biomechanical remodeling in aging and genetic mutant murine mitral valve leaflets: insights into Marfan syndrome. PLoS ONE. 2012;7:e44639. doi: 10.1371/journal.pone.0044639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.deAlmeida A, McQuinn T, Sedmera D. Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ Res. 2007;100:1363–1370. doi: 10.1161/01.RES.0000266606.88463.cb. [DOI] [PubMed] [Google Scholar]
- 32.Sedmera D, Pexieder T, Rychterova V, Hu N, Clark EB. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat Rec. 1999;254:238–252. doi: 10.1002/(SICI)1097-0185(19990201)254:2<238::AID-AR10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 33.Wozniak MA, Chen CS. Mechanotransduction in development: a growing role for contractility. Nat Rev Mol Cell Biol. 2009;10:34–43. doi: 10.1038/nrm2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petroll WM, Ma L, Jester JV. Direct correlation of collagen matrix deformation with focal adhesion dynamics in living corneal fibroblasts. J Cell Sci. 2003;116:1481–1491. doi: 10.1242/jcs.00357. [DOI] [PubMed] [Google Scholar]
- 37.Matthews HK, Marchant L, Carmona-Fontaine C, Kuriyama S, Larraín J, Holt MR, Parsons M, Mayor R. Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development. 2008;135:1771–1780. doi: 10.1242/dev.017350. [DOI] [PubMed] [Google Scholar]
- 38.Saito K, Ozawa Y, Hibino K, Ohta Y. FilGAP, a Rho/Rho-associated protein kinase-regulated GTPase-activating protein for Rac, controls tumor cell migration. Mol Biol Cell. 2012;23:4739–4750. doi: 10.1091/mbc.E12-04-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Migeotte I, Grego-Bessa J, Anderson KV. Rac1 mediates morphogenetic responses to intercellular signals in the gastrulating mouse embryo. Development. 2011;138:3011–3020. doi: 10.1242/dev.059766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura F. FilGAP and its close relatives: a mediator of Rho-Rac antagonism that regulates cell morphology and migration. Biochem J. 2013;453:17–25. doi: 10.1042/BJ20130290. [DOI] [PubMed] [Google Scholar]
- 41.Lim SM, Trzeciakowski JP, Sreenivasappa H, Dangott LJ, Trache A. RhoA-induced cytoskeletal tension controls adaptive cellular remodeling to mechanical signaling. Integr Biol (Camb) 2012;4:615–627. doi: 10.1039/c2ib20008b. [DOI] [PubMed] [Google Scholar]
- 42.Doye A, Mettouchi A, Bossis G, Clément R, Buisson-Touati C, Flatau G, Gagnoux L, Piechaczyk M, Boquet P, Lemichez E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell. 2002;111:553–564. doi: 10.1016/s0092-8674(02)01132-7. [DOI] [PubMed] [Google Scholar]
- 43.Heuzé ML, Lamsoul I, Baldassarre M, Lad Y, Lévêque S, Razinia Z, Moog-Lutz C, Calderwood DA, Lutz PG. ASB2 targets filamins A and B to proteasomal degradation. Blood. 2008;112:5130–5140. doi: 10.1182/blood-2007-12-128744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakamura F, Heikkinen O, Pentikäinen OT, Osborn TM, Kasza KE, Weitz DA, Kupiainen O, Permi P, Kilpeläinen I, Ylänne J, et al. Molecular basis of filamin A-FilGAP interaction and its impairment in congenital disorders associated with filamin A mutations. PLoS One. 2009;4:e4928. doi: 10.1371/journal.pone.0004928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duval D, Lardeux A, Le Tourneau T, Norris RA, Markwald RR, Sauzeau V, Probst V, Le Marec H, Levine R, Schott JJ, et al. Valvular dystrophy associated filamin A mutations reveal a new role of its first repeats in small-GTPase regulation. Biochim Biophys Acta. 2014;1843:234–244. doi: 10.1016/j.bbamcr.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiu YN, Norris RA, Mahler G, Recknagel A, Butcher JT. Transforming growth factor b, bone morphogenetic protein, and vascular endothelial growth factor mediate phenotype maturation and tissue remodeling by embryonic valve progenitor cells: relevance for heart valve tissue engineering. Tissue Eng Part A. 2010;16:3375–3383. doi: 10.1089/ten.tea.2010.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butcher JT, McQuinn TC, Sedmera D, Turner D, Markwald RR. Transitions in early embryonic atrioventricular valvular function correspond with changes in cushion biomechanics that are predictable by tissue composition. Circ Res. 2007;100:1503–1511. doi: 10.1161/CIRCRESAHA.107.148684. [DOI] [PubMed] [Google Scholar]
- 48.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:121–130. doi: 10.1161/ATVBAHA.112.300504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yalcin HC, Shekhar A, Rane AA, Butcher JT. An ex-ovo chicken embryo culture system suitable for imaging and microsurgery applications. J Vis Exp. 2010:2154. doi: 10.3791/2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.