

Abstract
Using high-pressure NMR spectroscopy and differential scanning calorimetry, we investigate the folding landscape of the mature HIV-1 protease homodimer. The cooperativity of unfolding was measured in the absence or presence of an active site inhibitor DMP323, for the active protease (PR), its inactive variant PRD25N and an extremely multi-drug resistant mutant, PR20. The individual fit of the pressure denaturation profiles gives rise to first order, ΔGNMR, and second order, ΔVNMR (the derivative of ΔGNMR with pressure) apparent thermodynamic parameters for each amide proton considered. Heterogeneity in the apparent ΔVNMR values reflects departure from an ideal cooperative unfolding transition. The narrow to broad distribution of ΔVNMR spanning the extremes from inhibitor-free PR20D25N to PR-DMP323 complex, and distinctively for PRD25N-DMP323 complex, indicated large variations in folding cooperativity. Consistent with this data, the shape of thermal unfolding transitions varies from asymmetric for PR to nearly symmetric for PR20, as dimer-inhibitor ternary complexes. Lack of structural cooperativity was observed between regions located close to the active site, including the hinge and tip of the glycine-rich flaps, and the rest of the protein. These results strongly suggest that inhibitor binding drastically decreases the cooperativity of unfolding by trapping the closed flap conformation in a deep energy minimum. To evade this conformational trap, PR20 evolves exhibiting a smoother folding landscape with nearly an ideal two-state (cooperative) unfolding transition. This study highlights the malleability of retroviral protease folding pathways by illustrating how the selection of mutations under drug pressure remodels the free-energy landscape as a primary mechanism.
Keywords: HIV protease, drug resistance, protein folding landscape, high-pressure NMR, calorimetry
Graphical abstract
INTRODUCTION
The HIV-1 protease is synthesized as part of a ~162 kDa Gag-Pol polyprotein precursor and is responsible for its own release at its termini from the precursor to form the mature enzyme (Fig. 1a). It promotes the controlled proteolysis of the viral Gag and Gag-Pol polyproteins into mature structural and functional proteins required for virus assembly, maturation and propagation1–5. The mature HIV-1 protease is a dimeric aspartyl protease composed of two identical polypeptides of 99 amino acids each. The dimer is held together through interface contacts at the N- and C-termini (residues 1–4 and 96–99), the active site (residues 25–27) and to a lesser extent, the glycine-rich flaps (residues 44–57)6,7 (Fig. 1b and c). The interface is stabilized upon binding an inhibitor or substrate, which makes contacts with residues in the active site cavity and induces a closed flap conformation. The mature protease is one of the primary targets for antiviral therapy and several potent protease inhibitors (PIs) have been designed to bind to the active site of the dimer at low nanomolar and picomolar affinities8,9. However, the success of these drugs is accompanied by the rapid emergence of drug resistant mutants, facilitated by the intrinsic error-prone replication of the viral reverse transcriptase10,11, to select for a functionally competent enzyme in the presence of PIs. To date, 15 sites for major mutations and 19–20 for minor mutations have been identified for all nine FDA approved PIs9,12.
Figure 1.

(a) Domain organization of the HIV-1 Gag-Pol polyprotein. Abbreviations: MA, matrix; CA, capsid; SP1, spacer peptide 1; NC, nucleocapsid; TFR, transframe region; PR, protease; RT, reverse transcriptase; RN, ribonuclease; IN, integrase; (b) Sequence alignment of mature proteases used in this study. A construct optimized for structural studies and a surrogate of the wild-type, namely PR, bears mutations Q7K, L33I, L63I, C67A, and C95A (shown in blue) to restrict autoproteolysis (red arrows) and avoid cysteine-thiol oxidation45. Introduced mutations common to PR and PR20 are Q7K, C67A, and C95A. The 19 mutations, both naturally occurring and selected under drug pressure, in PR20 are shown in black and red lettering, respectively. Dots denote identical residues. Highly conserved regions and regions where major drug-resistance mutations occur are highlighted in yellow and light blue, respectively46. Regions of natural variation are not highlighted. Major drug-resistance mutations are indicted by red asterisks (as defined in http:/hivdb.stanford.edu/cgi-bin/PIResiNote.cgi and reference 47). X-ray structure of (c) the optimized wild-type protease (PR) in a closed conformation (PDB id: 3BVB)26 with active site residues shown in stick representation and (d) the inhibitor-free PR20 dimer in a wide-open conformation (PDB ID: 3UF3)15. (d) The location of major drug-resistance mutations (DRMs) are shown on one subunit in red circles and naturally selected/compensatory mutations are shown on the other subunit in black on white circles. Highly conserved regions and regions where major drug-resistance mutations occur are shaded on the x-ray structure in yellow and light blue, respectively, matching the color scheme in (b) for those same regions.
A recently characterized multi-drug resistant clinical isolate, PR20, which bears 19 drug-resistance mutations and one engineered mutation (Q7K) to restrict autoproteolysis (self-degradation, Fig. 1b and Fig. S1), is a highly evolved drug resistant variant13,14. PR20 exhibits a dimer dissociation constant (Kdimer) of ~30 nM, which is >3-fold higher than for optimized wild-type protease (PR, Fig. 1b and 1c), and is catalytically competent with a similar turnover rate (kcat) and an ~13-fold higher Km for a chromogenic substrate corresponding to the capsid/SP1 cleavage site in the Gag polyprotein, relative to PR. It undergoes efficient precursor autoprocessing and cleaves the Gag polyprotein in the correct order at the various sites to produce the mature structural proteins albeit only at an ~4-fold slower rate than PR14. However, PR20 shows a drastically lower affinity for PIs by >3 orders of magnitude relative to PR13. Crystallographic analysis of PR20 showed altered inter-subunit interactions and an unusually wide separation of the two flexible flaps in the absence of inhibitors15–16 (Fig. 1d). Studies indicate that mutations of residues 35–37 in the hinge loop are responsible for the perturbed flap conformation15. Specifically, E35D mutation breaks the ion pair with R57, and M36I forms new hydrophobic interactions with two other mutated residues in PR20, I15V and I33F, thus altering the flap conformation15. The open flap conformations of PR20 as well as the expanded binding site of the inhibitor-bound closed form suggest possible approaches for modifying inhibitors to target extreme drug-resistant HIV. Recent backbone residual dipolar couplings measured for the N-H amide vectors confirmed that PR20 adopts a wide-open flap conformation in solution17. Using a combined NMR/MD approach we also showed that pressure perturbation induces the opening of the glycine-rich flaps of the inhibitor-free PR, while no such effect was detected for the inhibitor-free PR2018 at subdenaturing pressure, therefore confirming that drug-resistance mutations affect the open-closed flap equilibrium of the PR dimer.
While the effects of drug-resistance mutations on the structural properties and conformational dynamics of the protease have been intensively investigated15–20, very little is known about the effect of these mutations on PR folding and possible relevance as a molecular basis for drug resistance. The folding mechanisms have been examined in several experimental and molecular dynamics studies21–25 pertaining only to the wild type PR and not to the drug-resistant variants. A fundamental understanding of the folding/dimerization events and dynamics of the protease in its precursor and mature forms, both as a monomer and dimer, will be crucial for the development of folding and dimerization inhibitors. Such alternative inhibition strategies may be required to overcome the emergence of extremely resistant variants such as PR2013 or PR22 bearing 22 mutations9.
In the present study, we monitored by high-pressure NMR spectroscopy the complete unfolding of dimers of the active PR, the inactive PR, bearing the active site mutation D25N (termed PRD25N), as well as the drug resistant mutant, PR20D25N. The active site D25N mutation is commonly introduced to enhance data quality by abolishing autoproteolysis of the uninhibited dimer26. We showed recently that inhibitor-free PRD25N maintains a closed flap conformation in solution17. The degree of folding cooperativity of each protease construct was independently estimated from the heterogeneity in pressure perturbation of individual amide protons. The atomic resolution offered by NMR experiments provides an intrinsic multi-probe approach to assess the degree of protein folding cooperativity, which is otherwise difficult to characterize using techniques such as circular dichroism or fluorescence. The interpretation of heterogeneity in the residue-per-residue NMR signals upon unfolding is not limited to pressure perturbation and is very similar to the analysis of midpoints of thermal unfolding reported by Munoz and coworkers for the downhill protein BBL27,28. Pressure perturbation also presents the advantage to destabilize locally the folded state of proteins based on the amount of internal void volume or packing defects in the folded structure, in contrast to temperature or chemical denaturants, which act globally in proportion to the change in the degree of exposure of surface area upon unfolding29–31.
The high-pressure NMR experiments were complemented with differential scanning calorimetry (DSC) to examine the thermal unfolding cooperativity of the same protease constructs. Large differences in folding cooperativity were observed for PR, PRD25N and PR20D25N. The highest degree of heterogeneity was exhibited by PR bound to the symmetric inhibitor DMP32332, followed by the PRD25N-DMP323 ternary complex and finally, the inhibitor-free PRD25N. In contrast, the drug-resistant mutant PR20D25N, with or without inhibitor, shows a very high degree of folding cooperativity. These results indicate that in the presence of the inhibitor DMP323, the inhibitor-bound conformation is trapped in a deep minimum, which decreases the unfolding cooperativity of the mature PR dimer. Furthermore, our results suggest that in order to bypass this conformational trap, PR20 evolves to function on a smooth and minimally frustrated free energy landscape that populates a single dimer conformation with open flaps in the native state.
RESULTS
Reversible pressure denaturation of the mature protease dimer
The 1H-15N NMR spectra of all protease constructs recorded at 20 °C and atmospheric pressure show the typical well resolved and dispersed amide resonances characteristic of folded proteins (Fig. 2a and S1). The complete pressure-induced unfolding was monitored by recording 1H-15N TROSY spectra at 20 °C, at pressures ranging from 1 to 2500 bar. At elevated pressure, the unfolding is characterized by a slow exchange process where both the native and the unfolded cross peaks become visible (Fig. 2b, 2c and S1). We verified that pressure denaturation is fully reversible by observing a complete recovery of the original NMR spectrum (both chemical shifts and intensity) when the pressure was lowered back to 1 bar (Fig. 2d). The unfolding reversibility ensures a proper thermodynamic characterization of the process, which is problematic to asses by heat denaturation because of the partial aggregation of PR upon its unfolding at high temperature. Thus, high-pressure perturbation is a reliable fully reversible method for studying the unfolding-refolding equilibrium of the mature protease.
Figure 2.

1H-15N TROSY spectra of 150 μM mature PR20D25N dimer in 20 mM sodium phosphate, pH 5.7, recorded at 20 °C in (a) 1, (b) 900 and (c) 1500 bar of pressure, showing the slow exchange process characterizing the pressure-induced unfolding as the cross peaks of the native and those of the unfolded states are both visible at intermediate pressure. (d) A complete recovery of the folded spectrum was observed when the pressure was lowered back to 1 bar after a high-pressure cycle.
The pressure-induced unfolding of PR20D25N was monitored by NMR as described above, yielding residue-specific intensity profiles as a function of pressure that were well described by a simple two-state unfolding model (Fig. S2). The individual fit of the pressure denaturation profiles gives rise to first order, ΔG, and second order, ΔV (the derivative of ΔG with pressure) apparent thermodynamic parameters for each amide proton considered29,31. Considering that the exchange between the native state and the potential folding intermediates occurs on a fast-to-intermediate time scale relative to the time scale of the NMR experiments, we monitored the loss of cross peak height (reflecting more precisely the residue-to-residue variations of cross peak width) rather than cross peak volume for our intensity profiles. To distinguish those apparent thermodynamic parameters from the proper thermodynamic parameters derived from calorimetry experiments, the NMR-derived parameters are named ΔGNMR and ΔVNMR in the present study.
The distribution of residue-specific apparent ΔVNMR values obtained for the PR20D25N dimer appears narrow and symmetric with an average ΔVNMR of 130.6 ml/mol (Fig. 3a and b). The pressure unfolding experiments recorded under the same conditions for the inhibitor-free PRD25N dimer, reveal a broader distribution of apparent ΔVNMR values, with a distribution width of 33.9 ml/mol, compared to 15 ml/mol for PR20D25N, and a smaller average ΔVNMR (109.3 ml/mol) (Fig. 3d and e). Both the broader distribution of the ΔVNMR values and the smaller average ΔVNMR indicate significantly less cooperative unfolding of the inhibitor-free PRD25N compared to PR20D25N. We also characterized the pressure-induced unfolding of dimer-inhibitor ternary complexes of PR and PRD25N. The cyclic non-peptide urea inhibitor DMP32332 was chosen because its 2-fold symmetry reduces the complexity of the NMR spectra as compared to an asymmetric inhibitor. In the presence of DMP323, PRD25N shows a very large heterogeneity in the individual amide proton intensity profiles (Fig. 3g and S2c) resulting in a clear double distribution of apparent ΔVNMR values, with one maximum at 103.4 ml/mol and a second maximum at 58.3 ml/mol (Fig. 3g and h). To address the question of the effect of inhibitor affinity on the unfolding cooperativity, we examined the pressure-induced unfolding of the inhibitor-bound active PR, which shows ~five times higher affinity for DMP323 compared to the inactive PRD25N 33. Compared to PRD25N, PR-DMP323 complex shows a single broad distribution of apparent ΔVNMR values with an average of only 39.2 ml/mol (Fig. 3j and k), indicating a very uncooperative unfolding transition. The magnitude of ΔVNMR values are shown on a ribbon representation of the corresponding 3-D structure for all of the samples analyzed (Fig. 3c, 3f, 3i and 3l).
Figure 3.

The ΔVNMR values calculated from the individual fit of the intensity as a function of pressure for each cross peak are shown in relation to the residue number for (a) PR20D25N (d) PRD25N, (g) PRD25N-DMP323 complex and (j) PR-DMP323 complex. The location of the nine major drug resistance mutations are indicated for PR20D25N with dashed vertical lines and red circles on top in (a). Distribution of calculated ΔVNMR values for (b) PR20D25N, (e) PRD25N, (h) PRD25N-DMP323 and (k) PR-DMP323. The magnitude of ΔVNMR values are mapped on the corresponding protease x-ray structure for (c) PR20D25N (PDB ID: 3UF315), (f) PRD25N, (i) PRD25N-DMP323 and (l) PR-DMP323 (PDB ID: 3BVB26). Residues for which the ΔVNMR value could not be accurately measured (including the proline residues) are shown in gray on the x-ray structures. The ensemble of cross peak 1H-15N intensity profiles as a function of pressure are shown in Fig. S2.
Using the complete sets of individual ΔVNMR and ΔGNMR values, we observed a strong correlation between these two apparent thermodynamic parameters for all four protease dimers (Fig. S3), suggesting that the magnitude of volume lost upon unfolding is correlated to the stability of the set of interactions locally affected. Interestingly, the slope of the ΔVNMR vs. ΔGNMR correlation varies considerably among the protease constructs, with the strongest correlation for the dimer with the most cooperative unfolding process, PR20D25N, and the weakest correlation for the least cooperative, the PR-DMP323 complex. For the latter, the large range of ΔGNMR, from 500 to 6500 cal/mol, correlates with a relatively small range of ΔVNMR, from 15 to 75 ml/mol, suggesting that the volume changes measured for this complex predominantly reflect the partial unfolding of different sets of interactions rather than the global unfolding of the dimer (Fig. S3).
Local destabilization of the dimeric structure
We then investigated whether the large heterogeneity observed by monitoring the loss of the folded dimer cross peaks as a function of pressure was due to a conformational heterogeneity of states corresponding either to the folded or the unfolded basin. The presence of conformational exchange on an intermediate or slow time scale within the unfolded basin can potentially be detected through the residue-to-residue variations of the unfolded cross peaks width or volume respectively as a function of pressure.
Using a standard combination of 3D NMR experiments, we fully assigned the backbone chemical shifts of the unfolded PRD25N at 2500 bar (Fig. S4 and Table S1) and used the assignment information to monitor the residue-specific increase of the unfolded cross peaks height as a function of pressure (reflecting the potential residue-specific variations of the cross peak widths or volumes). We found that the increase of the unfolded population is extremely cooperative with barely any heterogeneity in the pressure profile even for the residues showing the largest heterogeneity in the folded state (Fig. 4a and b). These results strongly suggest that lack of unfolding cooperativity observed for PRD25N in the presence of DMP323 is due to conformational exchange on a fast-to-intermediate time scale between multiple states within the folded basin.
Figure 4.

(a) Representative example of two sets of native contacts, R41-Q58 and V32-I54, identified from the reference structure of PRD25N (pdb id: 3BVB26), showing very different pressure dependence of their respective cross peak intensities. Based on the normalized cross-peak intensity, the probability of R41-Q58 to be maintained at 1000 bar is 94.7*96.9/100 = 91.8%, while the contact V32-I54 has much lower probability: 64.9*75.1/100=48.7% to be maintained at the same pressure. (b) Pressure profile measured for V32 and Q58 are shown for both the folded (solid line) and unfolded cross peaks (dashed line). (c) Fractional contact map at 1000 bar of PRD25N-DMP323 complex with the low probability (of maintaining contact) residues (p<50%) highlighted with large red circles (see Materials and Methods). The contacts between residues 1–99 and 100–198 depict the contacts formed at the dimer interface. (d) The network of native contacts that are the most pressure sensitive (p<50%) are represented in red on the PRD25N structure (PDB id: 3BVB26) and residue positions are labeled.
To narrow down the origin of the heterogeneity in the tertiary fold of the dimer, we constructed fractional contact maps at each pressure from the product of the normalized cross peak intensities of the two residues involved in each native contact of the dimer (see legend of Fig. 4). As previously described29,34, these contact maps reflect the probability that a given contact between two residues is maintained upon unfolding at a given pressure. The contact map derived from the experimental data recorded at 1000 bar for the PRD25N-DMP323 complex shows an interesting partitioning of the most pressure sensitive contacts (p < 50%) around the active site (highlighted in red in Fig. 4c and d), including mainly contacts between residues of the “hinge” region (residues 32, 33, 78, 82 and 83) and residues at the tip of the flaps (residues 47–50, 52 and 54–56). Such distinct localization of the most pressure-sensitive contacts around the binding site of DMP323 suggests that the uncooperativity of unfolding observed in our NMR experiments is due to the local conformational dynamics and partial unfolding occurring at intermediate pressures in the hinge and flap regions. Contact maps derived for PR20D25N, PRD25N and PR-DMP323 complex lack the same distribution of p <50% (Fig. S5) as seen for PRD25N-DMP323 complex.
Monitoring unfolding of protease dimer by DSC
NMR unfolding experiments provide an “atom-by-atom” description of the folding process but the analysis of the unfolding profiles involves several assumptions, such as the quasi-independence of the individual amide proton signals or the two-state fit of the individual unfolding curves29,31. In contrast to NMR, DSC experiments directly detect the heat absorbed during denaturation and provide an unbiased and model-free estimation of the unfolding cooperativity. The non-cooperative behavior is typically detected through the asymmetry of the DSC thermograms, which implies that the van’t Hoff enthalpy is smaller than the calorimetric enthalpy35. Combination of NMR and DSC experiments constitutes a powerful approach to examine both local and global aspects of protein folding thermodynamics that has been recently applied to the rapidly folding PBX homeodomain36.
To complement our high-pressure NMR experiments, we examined whether a non-cooperative behavior can also be verified from thermal unfolding. Consistent with the NMR pressure data (Fig. 3b), the almost symmetric DSC thermogram suggests a uniform 2-state unfolding transition for PR20D25N (Fig. 5a). The thermogram of PRD25N is less symmetric (Fig. 5b) and the asymmetry becomes increasingly pronounced for the DMP-bound forms of PRD25N and PR (Fig. 5c and d). The trailing upslope can be explained by the existence of partially unfolded species before and around the transition mid-point. The more pronounced asymmetry of the DSC thermogram observed for the DMP323-bound PRD25N compared to the inhibitor-free PRD25N (Fig. 5b and c) suggests that the presence of the inhibitor significantly decreases the unfolding cooperativity of the protease dimer, in good agreement with the NMR data. A distinct peak preceding the major transition appears in the thermogram when using DRV which exhibits a binding affinity of ~5 pM to PR (Fig. 5e), significantly stronger in binding than DMP323 (3.8 nM)32. In spite of a reasonably tight binding affinity of ~40 nM of DRV to PR2013, PR20-DRV complex still displays a nearly identical shape of the transition as that of inhibitor-free PR20D25N consistent with a cooperative behavior of unfolding (Fig. 5f). A DSC scan of PR20D25N in the presence of DMP323 did not change the transition midpoint of the DSC trace because of very weak binding consistent with the absence of observable chemical shift changes by NMR even when >2-fold molar excess of DMP323 was added to 150 μM PR20D25N dimer (data not shown). The very weak binding affinity of DMP323 to PR20D25N is a cumulative effect of the drug-resistance and D25N mutations as described13,26.
Figure 5.

DSC thermograms of (a) PR20D25N, (b) PRD25N, (c) PRD25N + DMP323, (d) PR + DMP323, (e) PR + DRV and (f) PR20 + DRV, all acquired using 15–40 μM protein (as dimer) maintained in 50 mM acetate buffer at pH 5.0. The curve in red and blue represents the deconvolution of a transition at a lower and higher Tm, respectively, using PeakFit 4.12 (Systat Software Inc., San Jose, CA). (e) Previously published DSC trace of PR-DRV complex is shown solely for the purpose of comparison26. In the absence of NMR pressure data, we speculated that the pre-transition peak could arise from the two binding orientations of DRV in the PR active site24, although our present findings indicate that this low temperature peak results from a non-two state (uncooperative) unfolding process.
Dimer-monomer equilibrium under pressure
As high-pressure is well known to induce the dissociation of oligomeric proteins37, we also examined the effect of pressure on dimer dissociation (Kdimer). We used the folding conditions optimized for the study of the monomeric protease38 to prepare inhibitor-free PRD25N and recorded 2D 1H-15N NMR spectra at a final dimer concentration of 14 and 75 μM. As the dimer-monomer equilibrium of the mature protease occurs on a slow time scale, both the dimer and monomer cross peaks were simultaneously monitored as a function of pressure (Fig. S6). These experiments are very similar to those performed by Ishima et al.38 for the chemical denaturation of ~10 μM dimer of PRD25N.
The intensities of four residues (G16, G52, A67 and G68) were averaged at each pressure to simultaneously monitor the pressure dependence of the three species (dimer, monomer and unfolded states). We observed a balance between the unfolding of the monomer and the dissociation of the dimer into monomers explaining the almost constant population of monomeric species over a large pressure range (ca. 1400 bar) (Fig. 6a and b). These unfolding experiments conducted at a lower protein concentration allowed us to determine both the Kdimer at atmospheric pressure and the volume change associated with the change of Kdimer with pressure, the apparent ΔVdiss. A Kdimer of 1.4 ± 0.2 μM was estimated from these experiments, in very good agreement with previously reported value of 1.3 μM for PRD25N 26, together with a ΔVdiss of −12 ± 4 ml/mol, which is significantly smaller than the ΔVdiss=−32.5 ml/mol recently reported by Ingr et al.39. The difference in the sample used (PRD25N instead of PR) and the experimental method (NMR instead of Trp fluorescence) likely explain the discrepancies in the estimated ΔVdiss values.
Figure 6.

Pressure denaturation profiles of inhibitor-free PRD25N monitored by acquiring 1H-15N HSQC spectra at (a) 75 μM and (b) 14 μM dimer in 50 mM sodium acetate, pH 5. Under these conditions, the individual cross peaks of both the monomer and dimer can be simultaneously monitored (Fig. S4). The cross peak volumes of 4 residues (G16, G52, A67, G68) were averaged at every pressure to plot these curves. These 4 particular residues were chosen because their corresponding cross peaks are perfectly resolved, without overlap, in all three states (dimer, monomer, unfolded).
DISCUSSION
The individual analysis monitoring the loss of folded cross peak intensities as a function of pressure according to a two-state model, for nearly every single backbone amide proton, gives rise to a distribution of apparent ΔVNMR values for each construct studied here. Fundamentally, a ΔV value represents the difference in volume between the folded and unfolded states and is derived from the change of the folding free energy, ΔGf, with pressure (p) : ΔVf = ∂ ΔGf/∂p = −ΔVu. Fitting spectroscopic observables, such as fluorescence or circular dichroism to two-state unfolding models is a common practice in urea, temperature and pressure perturbation studies. Certainly, for an ideally cooperative two-state unfolding transition, the cross peak intensity curves for all residues should converge toward a single ΔVNMR value, within experimental uncertainty. Thus, heterogeneity in the apparent ΔVNMR values measured for a given protein reflects departure from an ideal cooperative unfolding transition, and informs on the potential presence of intermediate states in the unfolding pathway29,31. In addition, comparison of the pressure profiles for the folded cross peaks and unfolded cross peaks indicates that such intermediates are present in the folded basin rather than in the unfolded basin (Fig. 4A and B).
While the multidrug resistant PR20D25N dimer showed narrow distributions of ΔVNMR values indicative of cooperative unfolding transitions, the observed distributions were much broader than the experimental uncertainty for the inhibitor-free PRD25N and the DMP323-bound PR and PRD25N (Fig. 3). In the extreme case of the PRD25N-DMP323 complex, we even observed a clear double distribution of ΔVNMR values, reflecting the lack of structural cooperativity between the regions located close to the active site (including the hinge region and the tip of the glycine-rich flaps) and the rest of the protein. Closely related to the width of the apparent ΔVNMR distributions, the average ΔVNMR also informs on the degree of unfolding cooperativity. For an ideal two-state cooperative transition, the difference in volume between the folded and unfolded states reflects the sum of solvent excluded cavities and void volume that are present in the folded structure and expelled upon unfolding29,30. However, if an intermediate state is populated upon unfolding, the average “apparent” ΔVNMR will be systematically underestimated compared to the real difference in volume between the folded and unfolded states. This is particularly clear for the PR dimer in the presence of DMP323 with an average ΔVNMR of only 39.2 ml/mol (Fig. 3k), which points to a very uncooperative unfolding transition.
The large variations in folding cooperativity observed for the four samples, spanning the extremes from the most cooperative (inhibitor-free PR20D25N) to the least (PR-DMP323), is also reflected in the change of the corollary relationship between the individual ΔVNMR and ΔGNMR values (Fig. S3). A very similar set of ΔVNMR vs. ΔGNMR correlations was also observed for the cavity-containing variants of SNase34 suggesting that the amount of volume lost in partial opening is generally correlated to the stability of the set of interactions locally affected. Correlations derived from pressure-induced unfolding experiments are closely related to the correlation between individual m values and ΔGx in denaturant-dependent H/D exchange experiments reported for RNase A40 and cytochrome c41. We found here that the slope of the ΔVNMR vs. ΔGNMR correlation is clearly related to the degree of folding cooperativity of the protease constructs and shows that in the extreme case of the DMP323-bound PR, a large range of interactions (from 500 cal/mol to 6500 cal/mol) can be disrupted without giving rise to a large volume change.
By comparing the pressure-induced unfolding of the inactive PRD25N dimer in the absence and presence of the inhibitor DMP323, we observed a clear decrease in the average apparent ΔVNMR and a large increase in the width of the ΔVNMR distribution (Fig. 3e and h). This demonstrates a significant decrease in the cooperativity of unfolding upon inhibitor binding. The fractional contact maps constructed at intermediate pressures for the PRD25N-DMP323 complex identify the contacts around the binding site of the inhibitor, including contacts formed at the tip of the flaps, to be the most pressure sensitive (Fig 4c and d). This spatial partitioning of the most pressure-sensitive contacts is consistent with the fact that cyclic urea inhibitors like DMP323 are known to stabilize the tip of the protease flaps by forming hydrogen bonds between the urea group oxygen and the amide proton of residues 50–50′32. The dissociation of the inhibitor appears as an obligatory preliminary step toward unfolding, leading to the overall decrease of unfolding cooperativity observed for PRD25N-DMP323 complex compared to the inhibitor-free PRD25N.
This decrease in the folding cooperativity upon inhibitor binding is also noticeable in calorimetric experiments. We found a clear increase in the asymmetry of the DSC thermograms for PRD25N in the presence of the DMP323, compared to the inhibitor-free PRD25N (Fig. 5). The affinity of DMP323 for the active PR is five times higher than for the inactive PRD25N 33 and, consistent with the NMR data, we also observed a much more asymmetric DSC thermogram for the more stable inhibitor-PR complex, the DMP323-bound PR, compared to DMP323-bound PRD25N. This effect was even more significant with the very high-affinity inhibitor darunavir (DRV). Reexamination of the DSC thermograms for the PRD25N dimer26, also clearly shows a less cooperative transition for the PRD25N dimer in complex with a substrate analogue inhibitor containing a reduced peptide bond as compared to the inhibitor-free PRD25N.
The pressure-induced unfolding experiments carried out on PR20D25N dimer show a narrow distribution of apparent ΔVNMR values, with a large average ΔVNMR of 130.6 mol/mol. These results clearly demonstrate that in contrast to the inhibitor-free PRD25N, PR20D25N unfolds in a highly cooperative manner, closely approximating an ideal two-state transition. In the absence of inhibitor at atmospheric pressure, PRD25N dimer was shown to adopt a closed conformation with very transient opening of the flaps42, while PR20D25N adopts mostly an open flap conformation in solution17,18. Based on the present results, we hypothesize that decreased cooperativity observed for unfolding of the inhibitor-free PRD25N reflects a three-state unfolding transition: closed-flap → open-flap → unfolded, while PR20D25N undergoes a simple two-state transition: open-flap → unfolded (Fig. 7).
Figure 7.

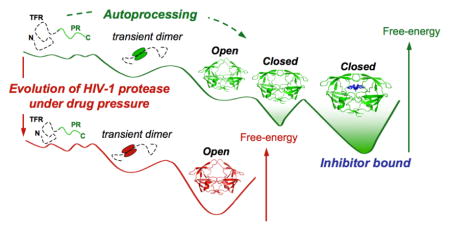
Schematic representation of the folding free energy landscape of HIV-1 protease before and after precursor autoprocessing and under drug pressure. The protease precursor is known to evolve on a very rugged energy landscape, populated predominantly by unfolded monomeric conformations45,48. Only a small fraction of active dimers are transiently formed initiating autoprocessing45,48. Optimal catalytic activity of the mature PR requires transitions between the closed and open flap conformers as a dimer (Fig. 3e). Addition of a competitive inhibitor blocks catalytic activity by trapping the bound conformation (with closed flaps) in a deep energy minimum (Fig. 3h and k). Selection of drug-resistance mutations smoothens the surface of the free-energy landscape, thus suppressing the conformational substates suitable to bind inhibitors. (Fig. 3b).
The increased cooperativity observed suggests that selection of drug-resistance mutations, typified by PR20, leads to a major remodeling of the folding free-energy landscape by selecting for the open flap, which presents the lowest affinity for inhibitors, among the two dimer conformations, the open and closed flaps. Overall, smoothening the surface of the free-energy landscape offers the advantage of suppressing local minima that could be potential targets for the binding of inhibitors. We therefore hypothesize that the major differences observed in folding cooperativity between the wild-type protease and the multidrug-resistant mutant PR20 illustrates the tenuous balance between the functional requirements for optimal catalytic activity (i.e. the equilibrium between the closed and open flap conformations) and the susceptibility to be trapped in a deep free-energy minimum upon inhibitor binding. Remarkably, the selection of a unique folded conformation with open flaps for the mature PR20 does not compromise the ability of this enzyme to undergo precursor autoprocessing and cleave the Gag polyprotein. Thus, inhibitors with moderate affinity, but with good specificity and bioavailability that target conserved regions essential for folding/dimerization may be effective because these conserved regions are essential for viability (i.e. structure/function) and are unlikely to accommodate the selection of resistance mutations under drug pressure.
To conclude, we monitored by NMR spectroscopy at a residue specific level, the pressure-induced unfolding of mature HIV-1 protease homodimer in the absence and presence of a symmetric inhibitor, DMP323, and compared the degree of unfolding cooperativity of the wild type protease with a recently characterized extremely drug resistant clinical isolate, PR20. We demonstrate that the DMP323-bound wild type protease is a trapped conformer occupying a deep minimum that increases the global roughness of the free energy landscape. PR20, on the other hand, functions on a smoother free energy landscape with a nearly ideal two-state unfolding transition, suggesting that evolutionary pressure selects against PR20-inhibitor traps. This study provides a direct illustration of the opposing dual requirement for optimal folding and catalytic function. In the case of HIV-1 protease, which is defined by a rugged energy landscape with two major dimer conformations involving opening and closing of the flaps for optimal function, selection of drug-resistance appears to involve a trade-off between a catalytically competent enzyme and a smooth energy landscape to avoid inhibitor binding.
MATERIALS AND METHODS
Sample preparation
Previously reported constructs6,13 used in this study are PR, PRD25N, and PR20. A new construct PR20D25N was created using the PR20 template, appropriate primers and Quik-change mutagenesis kit (Agilent Technologies, Santa Clara, CA). Uniformly 15N, 13C-labeled proteins were prepared by growing the cultures in minimal medium containing 15NH4Cl and 13C glucose as the sole nitrogen and carbon sources, respectively. Protease constructs were expressed and purified according to described protocols13. Briefly, proteins were fractionated from solubilized inclusion bodies under denaturing conditions in 4M guanidine hydrochloride, 50 mM Tris-HCl, pH 8, by size-exclusion chromatography (Superdex-75, GE Healthcare, Pittsburgh, PA) followed by reverse-phase HPLC (0.5 × 5 cm) on POROS 20 R2 resin (Life Technologies, Grand Island, NY) using a linear gradient from water/0.05% TFA to 60% acetonitrile/0.05% TFA over a period of 16 min at a flow rate of 4 mL/min. They were folded according to the schemes described38 for NMR and DSC experiments.
Differential scanning calorimetry
Proteins were freshly folded and concentrated to give a final dimer concentration of ~14 to 40 μM in 50 mM acetate buffer, pH 5. Constructs bearing the D25N mutation were scanned at ~40 μM concentrations. For experiments in the presence of inhibitors, a 2-fold molar excess of inhibitor relative to protease dimer concentration was added during folding. DSC scans were performed at a rate of 90 °C/h using a MicroCal VP-DSC microcalorimeter (Malvern, Westborough, MA) as previously described26 and raw data was processed using the Origin software provided with the instrument.
NMR
1H-15N TROSY-HSQC spectra were recorded on uniformly 15N/13C labeled samples using a 600 MHz Bruker Avance II spectrometer, equipped with a z-axis TCI cryogenic probe. A total of 200* × 1024* complex points were collected, for acquisition times of 104 and 121 ms in the 15N and 1H dimensions, respectively, using an interscan delay of 1.5 s. All the experiments were recorded using ~150 μM protein (as dimer) in 20 mM sodium phosphate, pH 5.7, at 20 °C. A commercial ceramic high-pressure NMR cell and an automatic pump system (Daedalus Innovations, Philadelphia, PA) were used to vary the pressure in the 1 bar to 2.5 kbar range. The peak height intensity profiles of all resolvable backbone amide cross-peaks as a function of pressure were fitted individually to a two-state unfolding model, yielding residue-specific estimates of the apparent free-energy difference (ΔGNMR) and volume changes (ΔVNMR) between the folded and the pressure-unfolded states. The list of native contacts was determined with a 4 Å cut-off distance from the reference x-ray structure 3BVB26. The probability of contact was then determined at each pressure from the product of the fractional intensities of the amide cross-peaks for the two residues involved in each native contact, as previously described29,34. The backbone assignment of the complete unfolded states of PRD25N at 2500 bar was based on 3D HNCO and 3D HNCACB spectra recorded at 600 MHz. The spectra were processed using NMRPipe43 and displayed with SPARKY44.
Supplementary Material
Highlights.
Drug resistance mutations affect the folding landscape of the mature HIV-1 protease.
The unfolding of the protease under pressure is fully reversible.
Binding of inhibitor decreases the folding cooperativity of the protease dimer.
The multidrug resistant PR20 exhibits a nearly ideal two-state unfolding transition.
Selection of drug resistance accounts for optimal folding and function.
Acknowledgments
We thank Hoi Sung Chung and Jane M. Sayer for discussions, Annie Aniana for technical assistance and Ad Bax for critical review of the work and support. This research was supported by the Intramural Research Program of the NIDDK, National Institutes of Health and the Intramural AIDS-Targeted Program of the Office of the Director, NIH. Clinical protease inhibitor daruvanir was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. We acknowledge the use of the NIDDK Advanced Mass Spectrometry Core Facility.
Abbreviations
- HIV-1
human immunodeficiency virus type 1
- PR
optimized wild-type mature HIV-1 protease
- PR20
extremely drug resistant variant of PR bearing 19 mutations
- PI
clinical protease inhibitor
- DRV
PI darunavir
- DMP323
cyclic nonpeptide urea inhibitor of PR
- DSC
differential scanning calorimetry
- NMR
nuclear magnetic resonance
- TROSY
transverse relaxation optimized spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oroszlan S, Luftig RB. Retroviral proteinases. Curr Top Microbiol Immunol. 1990;157:153–185. doi: 10.1007/978-3-642-75218-6_6. [DOI] [PubMed] [Google Scholar]
- 2.Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci USA. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplan AH, Manchester M, Swanstrom R. The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J Virol. 1994;68:6782–6786. doi: 10.1128/jvi.68.10.6782-6786.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Louis JM, Weber IT, Tozser J, Clore GM, Gronenborn AM. HIV-1 protease: maturation, enzyme specificity, and drug resistance. Adv Pharmacol. 2000;49:111–146. doi: 10.1016/s1054-3589(00)49025-3. [DOI] [PubMed] [Google Scholar]
- 5.Lee SK, Potempa M, Swanstrom R. The choregraphy of HIV-1 proteolytic processing and virion assembly. J Biol Chem. 2012;287:40867–40874. doi: 10.1074/jbc.R112.399444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louis JM, Ishima R, Torchia DA, Weber IT. HIV-1 Protease: Structure, dynamics and inhibition. Adv Pharmacol. 2007;55:261–298. doi: 10.1016/S1054-3589(07)55008-8. [DOI] [PubMed] [Google Scholar]
- 7.Todd MJ, Semo N, Freire E. The structural stability of the HIV-1 Protease. J Mol Biol. 1998;783:475–488. doi: 10.1006/jmbi.1998.2090. [DOI] [PubMed] [Google Scholar]
- 8.Shuman CF, Markgren PO, Hamalainen M, Danielson UH. Elucidation of HIV-1 protease resistance by characterization of interaction kinetics between inhibitors and enzyme. Antiviral Res. 2003;58:235–242. doi: 10.1016/s0166-3542(03)00002-0. [DOI] [PubMed] [Google Scholar]
- 9.Kozisek M, Henke S, Saskova KG, Jacobs GB, Schuch A, Buchholz B, Muller V, Krausslich HG, Rezacova P, Konvalinka J, Bodem J. Mutations in HIV-1 gag and pol compensate for the loss of viral fitness caused by a highly mutated protease. Antimicrob Agents Chemother. 2012;56:4320–4330. doi: 10.1128/AAC.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 11.Hu WS, Hughes SH. HIV-1 reverse transcription. Cold Spring Harb Perspect Med. 2012;2:1–22. doi: 10.1101/cshperspect.a006882. 006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber IT, Agniswamy J. HIV-1 Protease: structural perspectives on drug resistance. Viruses. 2009;1:1110–1136. doi: 10.3390/v1031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louis JM, Aniana A, Weber IT, Sayer JM. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitor. Proc Natl Acad Sci USA. 2011;108:9072–9077. doi: 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Louis JM, Deshmukh L, Sayer JM, Aniana A, Clore GM. Mutations proximal to sites of autoproteolysis and the α-helix that co-evolve under drug pressure modulate the autoprocessing and vitality of HIV-1 Protease. Biochemistry. 2015;54:5414–5424. doi: 10.1021/acs.biochem.5b00759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agniswamy J, Shen CH, Aniana A, Sayer JM, Louis JM, Weber IT. HIV-1 Protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochemistry. 2012;51:2819–2828. doi: 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen CH, Chang YC, Agniswamy J, Harrison RW, Weber IT. Conformational variation of an extreme drug resistant mutant of HIV protease. J Mol Graph Model. 2015;62:87–96. doi: 10.1016/j.jmgm.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roche J, Louis JM, Bax A. Conformation of inhibitor-free HIV-1 protease derived from NMR spectroscopy in a weakly oriented solution. Chembiochem. 2015;16:214–218. doi: 10.1002/cbic.201402585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roche J, Louis JM, Bax A, Best RB. Pressure-induces structural transition of mature HIV-1 protease from a combined NMR/MD simulation approach. Proteins. 2015;83:2117–2123. doi: 10.1002/prot.24931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai Y, Yilmaz NK, Myint W, Ishima R, Schiffer CA. Differential flap dynamics in wild-type and a drug resistant variant of HIV-1 protease revealed by molecular dynamics and NMR. J Chem Theory Comput. 2012;8:3452–3462. doi: 10.1021/ct300076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Y, Myint W, Paulsen JL, Schiffer CA, Ishima R, Yilmaz NK. Drug resistance mutations alter dynamics of inhibitor-bound HIV-1 Protease. J Chem Theory Comput. 2014;14:3438–3448. doi: 10.1021/ct4010454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy Y, Caflish A, Onuchic JN, Wolynes PG. The folding and dimerization of HIV-1 Protease: evidence for a stable monomer from simulations. J Mol Biol. 2004;340:67–79. doi: 10.1016/j.jmb.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 22.Broglia RA, Levy Y, Tiana G. HIV-1 protease folding and the design of drugs which do not create resistance. Curr Opin Struct Biol. 2008;18:60–66. doi: 10.1016/j.sbi.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 23.Verkhivker G, Tiana G, Camilloni C, Provasi D, Broglia RA. Atomistic simulation of the HIV-1 Protease folding inhibition. Biophys J. 2008;95:550–562. doi: 10.1529/biophysj.107.127621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noel AF, Bilsel O, Kundu A, Wu Y, Zitzewitz JA, Matthews RC. The folding free-energy surface of HIV-1 protease: insights into the thermodynamics basis for resistance to inhibitors. J Mol Biol. 2009;387:1002–1016. doi: 10.1016/j.jmb.2008.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rout MK, Reddy JG, Phillips M, Hosur RV. Single point mutation induced alterations in the equilibrium structural transitions on the folding landscape of HIV-1 protease. J Biomol Struct Dyn. 2013;31:684–693. doi: 10.1080/07391102.2012.707459. [DOI] [PubMed] [Google Scholar]
- 26.Sayer JM, Liu F, Ishima R, Weber IT, Louis JM. Effect of the active site D25N mutation on the structure, stability, and ligand binding of the mature HIV-1 protease. J Biol Chem. 2008;283:13459–13470. doi: 10.1074/jbc.M708506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadqi M, Fushman D, Munoz V. Atom-by-atom analysis of global downhill protein folding. Nature. 2006;442:317–321. doi: 10.1038/nature04859. [DOI] [PubMed] [Google Scholar]
- 28.Naganathan AN, Munoz V. Determining denaturation midpoints in multiprobe equilibrium protein folding experiments. Biochemistry. 2008;47:6752–6761. doi: 10.1021/bi800336x. [DOI] [PubMed] [Google Scholar]
- 29.Roche J, Caro JA, Norberto DR, Barthe P, Roumestand C, Schlessman JL, Garcia AE, Garcia-Moreno BE, Royer CA. Cavities determine the pressure unfolding of proteins. Proc Natl Acad Sci USA. 2012;109:6945–6950. doi: 10.1073/pnas.1200915109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rouget JB, Aksel T, Roche J, Saldana JL, Garcia AE, Barrick D, Royer CA. Size and sequence and the volume change of protein folding. J Am Chem Soc. 2011;133:6020–6027. doi: 10.1021/ja200228w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Oliveira GAP, Silva JL. A hypothesis to reconcile the physical and chemical unfolding of proteins. Proc Natl Acad Sci USA. 2015;112:E2775–84. doi: 10.1073/pnas.1500352112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ry Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang CH, Weber PC, Jackson DA, Sharpe TR, Erickson-Viitanen S. Rational design of potent, bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. Science. 1994;263:380–384. doi: 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- 33.Sayer JM, Louis JM. Interactions of different inhibitors with active-site aspartyl residues of HIV-1 protease and possible relevance to pepsin. Proteins. 2009;75:556–568. doi: 10.1002/prot.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roche J, Dellarole M, Caro JA, Guca E, Norberto DR, Yang Y, Garcia AE, Roumestand C, Garcia-Moreno BE, Royer CA. Remodeling of the folding free energy landscape of staphylococcal nuclease by cavity-creating mutations. Biochemistry. 2012;51:9535–9546. doi: 10.1021/bi301071z. [DOI] [PubMed] [Google Scholar]
- 35.Cortajarena AL, Regan L. Calorimetric study of a series of designated repeat proteins: modular structure and modular folding. Protein Sci. 2011;20:336–340. doi: 10.1002/pro.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farber P, Darmawan H, Sprules T, Mittermaier A. Analyzing protein folding cooperativity by differential scanning calorimetry and NMR spectroscopy. J Am Chem Soc. 2010;132:6214–6222. doi: 10.1021/ja100815a. [DOI] [PubMed] [Google Scholar]
- 37.Luong TQ, Kapoor S, Winter R. Pressure: A gateway to fundamental insights into protein solvation, dynamics and function. Chemphyschem. 2015 doi: 10.1002/cphc.201500669. [DOI] [PubMed] [Google Scholar]
- 38.Ishima R, Torchia DA, Louis JM. Mutational and structural studies aimed at characterizing the monomer of HIV-1 protease and its precursor. J Biol Chem. 2007;282:17190–17199. doi: 10.1074/jbc.M701304200. [DOI] [PubMed] [Google Scholar]
- 39.Ingr M, Lange R, Halabalova V, Yehya A, Hrncirik J, Chevalier-Lucia D, Palmade L, Blayo C, Konvalinka J, Dumay E. Inhibitor and substrate binding induced stability of HIV-1 protease against sequential dissociation and unfolding by high-pressure spectroscopy and kinetics. PLoS One. 2015;10:e0119099. doi: 10.1371/journal.pone.0119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayo SL, Baldwin RL. Guanidinium chloride induction of partial unfolding in amide proton exchange in SNase A. Science. 1993;269:873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 41.Bay Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freedberg DI, Ishima R, Jacob R, Wang YX, Kustanovich I, Louis JM, Torchia DA. Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 2002;11:221–232. doi: 10.1110/ps.33202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delaglio F, Grzesiel S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;5:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 44.Goddard TD, Kneller DG. Sparky 3. University of California San Francisco; San Francisco, CA: 2010. [Google Scholar]
- 45.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 46.Ceccherini-Silberstein F, Erba F, Gago F, Bertoli A, Forbici F, Bellocchi MC, Gori C, D’Arrigo R, Marcon L, Balotta C, Antinori A, Monforte AD, Perno CF. Identification of the minimal conserved structure of HIV-1 protease in the presence and absence of drug pressure. AIDS. 2004;18:F11–19. doi: 10.1097/01.aids.0000131394.76221.02. [DOI] [PubMed] [Google Scholar]
- 47.Wensing AM, Calvez V, Gunthard HF, Johnson VA, Paredes R, Pillay D, Shafer RW, Richman DD. Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 48.Tang C, Louis JM, Aniana A, Suh JY, Clore GM. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature. 2008;455:693–696. doi: 10.1038/nature07342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.