

Abstract
Objectives
Heat Shock Protein 90 (HSP90) is a molecular chaperone that stabilizes many oncogenic proteins. HSP90 inhibitors may sensitize tumors to cytotoxic agents by causing client protein degradation. Gemcitabine, which has modest activity in pancreas cancer, activates Chk1, a client protein of HSP90. This phase II trial was designed to determine whether 17AAG could enhance the clinical activity of gemcitabine through degradation of Chk1 in patients with stage IV pancreatic cancer.
Methods
A multicenter, prospective study combining gemcitabine and 17AAG enrolled patients with stage IV pancreatic adenocarcinoma, adequate liver and kidney function, ECOG performance status 0-2, and no prior chemotherapy for metastatic disease. The primary goal was to achieve a 60% overall survival at six months. Sixty-six patients were planned for accrual, with an interim analysis after 25 patients enrolled. Results: After a futility analysis to achieve the endpoint, accrual was halted with 21 patients enrolled. No complete or partial responses were seen. 40% of patients were alive at 6 months. Median overall survival was 5.4 months. Tolerability was moderate, with 65% of patients having ≥ grade 3 adverse events (AE), and 15% having grade 4 events.
Conclusions
The lack of clinical activity suggests that targeting Chk1 by inhibiting HSP90 is not important in pancreatic cancer sensitivity to gemcitabine alone. Further studies of HSP90 targeted agents with gemcitabine alone are not warranted.
Keywords: HSP90 inhibitor, 17AAG, gemcitabine, Chk1, pancreatic cancer, phase II
Introduction
Pancreas cancer remains a highly lethal malignancy with little improvement in survival gained from various therapies over the past half-century [1]. For instance, gemcitabine, a pyrimidine nucleoside analog that induces G1/S cell cycle arrest via increased Chk1 expression, as monotherapy for pancreatic cancer has modest activity [2]. As evidenced by its successful combination with nab-paclitaxel [3], gemcitabine could be a more effective drug if further synergistic combinations were found.
Heat shock protein 90 (HSP90) is a key chaperone protein responsible for stabilizing and maintaining the activity of multiple client proteins, such as Chk1, akt, mutant p53, Cdk4, and erbB2, that are potentially involved in the cell cycle dysregulation and results in pancreatic carcinogenesis, [4-10]. 17-N-Allylamino-17-demethoxygeldanamycin (Tanespimycin/17AAG) is a geldanamycin-analog HSP90 inhibitor that leads to increased ubiquitin-mediated degradation of its client proteins. Gemcitabine inhibits DNA synthesis leading to premature chain termination during replication. This leads to activation of Chk1 and subsequent cell-cycle arrest, which can prevent cell death and increase survival [11]. Chk1 is a serine-threonine kinase that functions to induce cell cycle arrest at S-phase in response to DNA damage [12]. Arlander et al. [12] demonstrated that 17AAG targeting of HSP90 led to Chk1 degradation, thus enhancing the cytotoxicity of gemcitabine. In view of the 17AAG and gemcitabine synergy demonstrated in vitro [12], the clinical activity of the combination in patients with metastatic pancreatic cancer was evaluated. Previously we found that the maximally tolerated doses of gemcitabine and tanespimycin on a 21 day cycle were 750 mg/m2 on days 1 and 8 and 154 mg/m2 on days 2 and 9 respectively [13]. We report the results of a phase II multicenter trial of 17AAG and gemcitabine using these doses and schedule of administration.
Methods
Patient Selection and Characteristics
Patients were required to have histologically or cytologically confirmed metastatic pancreatic adenocarcinoma, were ≥ 18 years old, had a life expectancy of ≥ 12 weeks, ECOG performance score of 0, 1, or 2, absolute neutrophil count > 1500, platelet count ≥100,000, total bilirubin within institutional upper limit of normal (ULN), AST ≤ 2.5× ULN, alkaline phosphatase ≤ 2× ULN (unless liver metastases present when up to ≤ 5× ULN), and creatinine within normal range. Patients had to be able and willing to sign and understand written consent, and to use adequate birth control methods if of reproductive age.
Patients using concurrent chemotherapy, having a history of allergic reactions to gemcitabine or 17AAG, having uncontrolled intercurrent illness, having prior radiation to the heart, being predisposed to cardiac arrhythmias or heart failure were excluded. No prior therapy for metastatic disease was allowed. Adjuvant therapy or therapy for locally advanced disease was allowed if greater than three months prior to enrollment. Prior radiation must have been completed three weeks prior to registration.
Trial Design
Eligible patients were given combination 17AAG (154 mg/m2, National Cancer Institute Investigational Drug Branch, Bethesda, MD) and gemcitabine (750 mg/m2) over 21 day cycles. All patients received gemcitabine on days 1 and 8 with 17AAG on days 2 and 9. Treatment was continued until evidence of disease progression, unacceptable adverse events, development of a significant comorbid condition, or patient withdrawal occurred.
The primary outcome was survival at six months, with secondary endpoints of overall survival, progression free survival, response rate, and toxicity. When noting no responses, a futility analysis was performed after accrual of 20 patients.
Statistical Methods
Historically, the 6-month survival for patients treated with gemcitabine alone is approximately 46% [2]; therefore, improving the survival rate to 60% or more was deemed to be of clinical interest and considered sufficiently promising to explore further.
All patients who met the eligibility criteria, signed a consent form, and began treatment were considered evaluable for the primary endpoint. This study design had a 90% probability of concluding that the regimen was promising if the true success rate was 60%, at a 5% level of significance. Per study design, an interim analysis was to be performed after the first 25 evaluable patients entered the trial. If at least 12 patients lived for 6 months or longer, the study would then proceed to the full accrual of 66 evaluable patients. At the final analysis, if 33 or more patients (50%) lived at least 6 months, this would be considered adequate evidence of promising activity, and would warrant further testing of this regimen in subsequent studies. A confidence interval for the 6-month survival rate was calculated using the exact binomial method. After only 21 patients were enrolled, a futility analysis was undertaken and, as the endpoint could not be reached, the trial was summarily closed.
Secondary endpoints included adverse events, the confirmed response rate, progression-free survival, and overall survival. Adverse events were summarized in a tabular manner as the maximum grade for a given type of event for each patient. The commonly occurring grade 3+ adverse events are also reported. Kaplan-Meier methodology [14] was used to describe the distributions of progression-free and overall survival.
Results
Baseline Characteristics
Between May of 2008 and September of 2010, 21 patients were enrolled from the Mayo Clinic and Washington University in St. Louis and the characteristics are summarized in Table 1. Of these 21 patients, 20 were evaluable for analysis (one patient withdrew prior to receiving any treatment). The median age was 61.5 years (range: 51-81), the majority of patients were men (55%), nearly all had an ECOG PS of 0 or 1 (95%), and most did not have any prior adjuvant therapy.
Table 1.
Patient baseline demographics.
| Total (N=20) | |
|---|---|
|
| |
| Age | |
| Median | 61.5 |
| Range | (51.0-81.0) |
| Gender | |
| Female | 9 (45.0%) |
| Male | 11 (55.0%) |
| Ascites Present | |
| Yes | 2 (10.0%) |
| No | 18 (90.0%) |
| Performance Score | |
| 0 | 10 (50.0%) |
| 1 | 9 (45.0%) |
| 2 | 1 (5.0%) |
| Race | |
| White | 19 (95.0%) |
| Black or African American | 1 (5.0%) |
| Prior Surgery | |
| Yes | 8 (40.0%) |
| No | 12 (60.0%) |
| Previous Radiation Therapy | |
| Yes | 2 (10.0%) |
| No | 18 (90.0%) |
| Prior Therapy | |
| Yes | 2 (10.0%) |
| No | 18 (90.0%) |
Outcome measures
Of the 20 patients evaluable for the outcome measures of survival, progression-free survival, and response (Table 2), all have died and 16 (80%) had a documented disease progression prior to expiration. Eight of the 20 patients (40%) survived at least six months (95% CI: 19% to 64%). After 21 accruals, there was a <10% probability that the goal of improved six-month survival rate could be achieved with only five patients left to accrue. Given the 40% six-month survival rate in the first 20 patients, the trial was concluded early.
Table 2.
Patient disease responses with 17AAG and gemcitabine therapy. Objective responses by RECIST criteria are reported.
| Clinical Outcome | Total (%)* |
|---|---|
|
| |
| Objective Response | |
| Complete Response | 0 (0) |
| Partial Response | 0 (0) |
| Stable Disease | 10 (53) |
| Progression | 9 (47) |
One patient did not have a post-baseline tumor assessment and was excluded.
The median survival (Table 2; Figure 1A) was 5.4 months (95% CI: 3.1 to 7.7 months) and the median progression-free survival (Table 2; Figure 1B) was 2.6 months (95% CI: 1.4 to 4.0 months). There were no confirmed responses to 17AAG/gemcitabine therapy, although ten patients had a best clinical response of stable disease.
Figure 1.
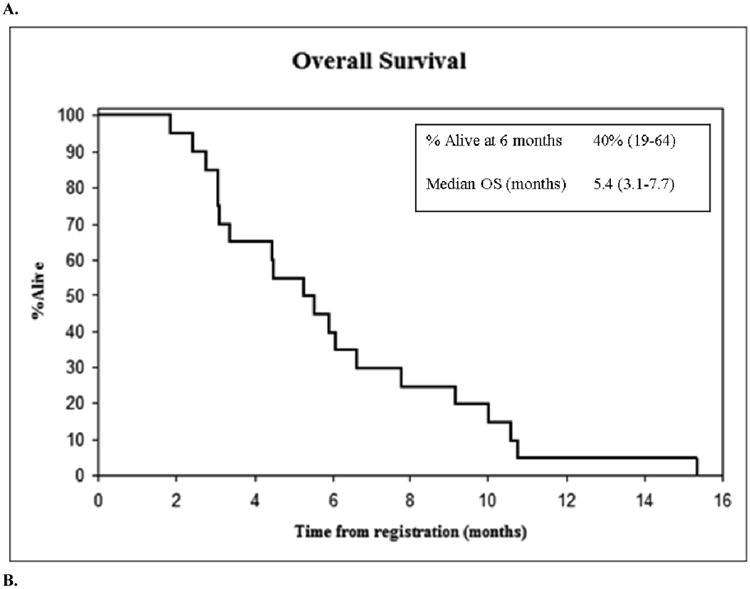
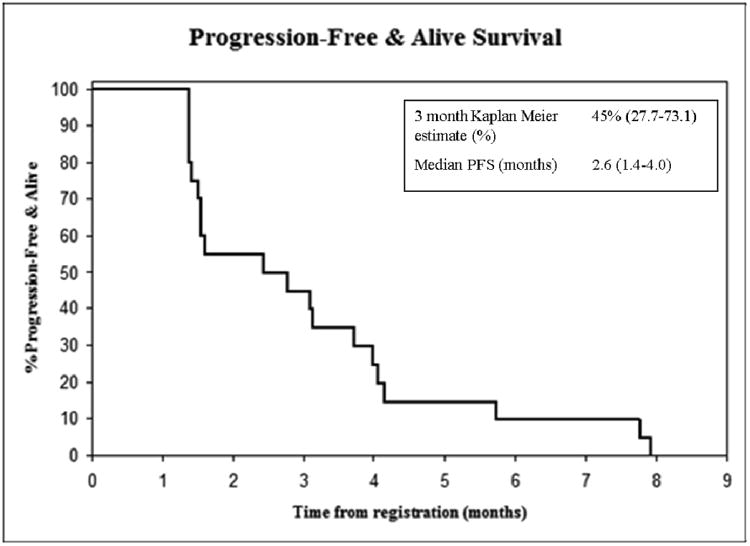
(A) Overall survival and (B) progression free survival of 20 evaluable patients by Kaplan-Meier estimation.
Adverse Events
Twenty patients were evaluable for adverse events (AE). Across all patients with adverse events at least “possibly” related to therapy, 13 (65%) experienced at least one grade 3 or worse AE, and two patients had a grade 4 AE (Table 3): one with grade 4 neutropenia and one with grade 4 lymphopenia (possibly related to treatment). No patients experienced a grade 5 AE. Commonly occurring Grade 3 adverse events included: nausea (15%), vomiting (15%), dehydration (10%), constipation (10%), anorexia (10%), lymphopenia (10%), and leukopenia (10%).
Table 3.
Commonly Occurring Maximum Severity (Grade 3/4) Adverse Events across all cycles of treatment (at least “possibly” attributed to therapy). There were no treatment related deaths.
| NCI CTC CATEGORY * | Frequency (%) (N=20) | |
|---|---|---|
|
| ||
| Grade 3 | Grade 4 | |
|
| ||
| Hematologic | ||
| Lymphopenia | 2 (10) | 1 (5) |
| Leukopenia | 2 (10) | 0 (0) |
| Neutropenia | 1 (5) | 1 (5) |
|
| ||
| GI | ||
| Nausea | 3 (15) | 0 (0) |
| Vomiting | 3 (15) | 0 (0) |
| Anorexia | 2 (10) | 0 (0) |
| Constipation | 2 (10) | 0 (0) |
| Dehydration | 2 (10) | 0 (0) |
| Dyspepsia | 1 (5) | 0 (0) |
| Increased alanine aminotransferase | 1 (5) | 0 (0) |
| Increased alkaline phosphatase | 1 (5) | 0 (0) |
|
| ||
| Miscellaneous | ||
| Thrombosis | 1 (5) | 0 (0) |
| Peripheral edema | 1 (5) | 0 (0) |
| Asthenia | 1 (5) | 0 (0) |
| Abdominal pain | 1 (5) | 0 (0) |
| Fatigue | 1 (5) | 0 (0) |
| Rash | 1 (5) | 0 (0) |
| Syncope | 1 (5) | 0 (0) |
NCI CTC Version 3.0
Of the 20 patients receiving treatment, 15 (75%) discontinued early due to disease progression, and five (25%) refused further treatment. A median of two cycles of therapy were given (range: 1- 8). All patients received the full dose of gemcitabine and 17AAG during cycle 1. From cycles 2-6, the percentage of patients receiving the full dose varied from 33% to 68% for gemcitabine and 25% to 60% for 17AAG.
Discussion
This phase II multicenter study of gemcitabine chemotherapy in combination with 17AAG, an HSP90 inhibitor, unfortunately did not result in significantly increased six-month, overall, or progression-free survival over expected in an interim analysis of 20 patients with metastatic pancreas adenocarcinoma. This result is consistent with studies of 17AAG in other malignancies, including acute leukemia and ovarian cancer [15,16].
Preclinical studies showed that, while 17AAG appears to decrease levels of Chk1 leading to an absence of S-phase checkpoint inhibition and synergistic cytotoxicity with gemcitabine in a dose-dependent manner, clinical trials have not borne out similar results for 17AAG combinations [15,16]. This could indicate that Chk1 is a clinically relevant marker, but when proteins such as akt, Chk1, and Raf have been isolated from patients' mononuclear cells and densitometrically assessed, there has often been only modest decreases in expression and this effect is transient, suggesting that targets are not affected long enough to see clinical benefit. Additionally, Chk1 may not be the most important mechanism of resistance. If degradation, nuclear transport, or other activation processes play a larger role in limiting gemcitabine activity, then HSP90 inhibition would play a lesser role in improving treatment efficacy. Another potential explanation for the lack of clinical response in this combination therapy is the activity of heat shock factor 1 (HSF1), a transcription factor activated during the heat shock response and when HSP90 interacts with the parent drug of 17AAG, geldanamycin [17-21]. HSF-/- knockout mice show increased 17AAG inhibition of HSP90 activity compared to normal mice [22]. As the gemcitabine/17AAG combination does not block HSF activity, there may be attenuation of its clinical benefit via the HSF1-induced protective mechanism.
Additional limitations in the efficacy of 17AAG in the clinic are secondary to the drug's toxicity in this combination. In our study, the majority of patients experienced grade 3 or higher adverse events. While phase I data suggest that the drug is well tolerated in monotherapy, even at higher dose levels than used in this trial [23,13,16], only one was carried out in combination with gemcitabine which showed similar tolerance to this regimen. As a result, the doses of both gemcitabine and 17AAG had to be dose reduced compared to the maximally tolerated doses in monotherapy. This could have conceivably led to the lower than expected response to the combination. While the goal of combined therapy was synergistic cytotoxicity, this was not achieved in this trial. There is certainly still the opportunity; however, that newer, less toxic HSP90 inhibitors [24] may be more effective if toxicity can be limited and doses or target effects maximized [25-27].
In conclusion, this phase II clinical trial supports the hypothesis that 17AAG can be administered in combination with gemcitabine in patients with pancreatic adenocarcinoma; however, there is a steep increase in toxicity over what would be expected for gemcitabine alone without any evident benefit in outcomes. Whether the failure is a result of this particular HSP90 inhibitor's limitations or of the approach as a whole has yet to be determined. With our finding, other HSP90 inhibitory agents in treatment of pancreatic cancer should be pursued with caution.
Acknowledgments
funding disclosure: Supported by P50 SPORE CA102701, N01-CM-2011-00099, CA15083, Georgeson Professorship Fund
Contributor Information
Katrina S. Pedersen, Division of Medical Oncology, Mayo Clinic, Rochester, Minnesota.
George P. Kim, Department of Biomedical Statistics and Informations, Mayo Clinic, Rochester, Minnesota.
Nathan R. Foster, Department of Biomedical Statistics and Informations, Mayo Clinic, Rochester, Minnesota.
Andrea Wang-Gillam, Division of Oncology, Washington University, Saint Louis, Missouri.
Charles Erlichman, Division of Medical Oncology, Mayo Clinic, Rochester, Minnesota.
Robert R. McWilliams, Division of Medical Oncology, Mayo Clinic, Rochester, Minnesota.
References
- 1.Society AC. Cancer Facts and Figures 2013. American Cancer Society; Atlanta: 2013. [Google Scholar]
- 2.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18(3):306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 5.An WG, Schnur RC, Neckers L, Blagosklonny MV. Depletion of p185erbB2, Raf-1 and mutant p53 proteins by geldanamycin derivatives correlates with antiproliferative activity. Cancer Chemother Pharmacol. 1997;40(1):60–64. doi: 10.1007/s002800050626. [DOI] [PubMed] [Google Scholar]
- 6.Schnur RC, Corman ML, Gallaschun RJ, Cooper BA, Dee MF, Doty JL, Muzzi ML, Moyer JD, DiOrio CI, Barbacci EG, et al. Inhibition of the oncogene product p185erbB-2 in vitro and in vivo by geldanamycin and dihydrogeldanamycin derivatives. J Med Chem. 1995;38(19):3806–3812. doi: 10.1021/jm00019a010. [DOI] [PubMed] [Google Scholar]
- 7.Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, Chung EJ, Trepel J, Neckers L. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res. 2003;63(22):7777–7784. [PubMed] [Google Scholar]
- 8.Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Skele KL, Hoffman JP, Testa JR. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2002;87(4):470–476. doi: 10.1002/jcb.10287. [DOI] [PubMed] [Google Scholar]
- 9.Semba S, Moriya T, Kimura W, Yamakawa M. Phosphorylated Akt/PKB controls cell growth and apoptosis in intraductal papillary-mucinous tumor and invasive ductal adenocarcinoma of the pancreas. Pancreas. 2003;26(3):250–257. doi: 10.1097/00006676-200304000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8(1):27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 11.Karnitz LM, Flatten KS, Wagner JM, Loegering D, Hackbarth JS, Arlander SJ, Vroman BT, Thomas MB, Baek YU, Hopkins KM, Lieberman HB, Chen J, Cliby WA, Kaufmann SH. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68(6):1636–1644. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 12.Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem. 2003;278(52):52572–52577. doi: 10.1074/jbc.M309054200. [DOI] [PubMed] [Google Scholar]
- 13.Hubbard J, Erlichman C, Toft DO, Qin R, Stensgard BA, Felten S, Ten Eyck C, Batzel G, Ivy SP, Haluska P. Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Invest New Drugs. 2011;29(3):473–480. doi: 10.1007/s10637-009-9381-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaplan E, Meier P. Nonparametric estimation for incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 15.Hendrickson AE, Oberg AL, Glaser G, Camoriano JK, Peethambaram PP, Colon-Otero G, Erlichman C, Ivy SP, Kaufmann SH, Karnitz LM, Haluska P. A phase II study of gemcitabine in combination with tanespimycin in advanced epithelial ovarian and primary peritoneal carcinoma. Gynecol Oncol. 2012;124(2):210–215. doi: 10.1016/j.ygyno.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaufmann SH, Karp JE, Litzow MR, Mesa RA, Hogan W, Steensma DP, Flatten KS, Loegering DA, Schneider PA, Peterson KL, Maurer MJ, Smith BD, Greer J, Chen Y, Reid JM, Ivy SP, Ames MM, Adjei AA, Erlichman C, Karnitz LM. Phase I and pharmacological study of cytarabine and tanespimycin in relapsed and refractory acute leukemia. Haematologica. 2011;96(11):1619–1626. doi: 10.3324/haematol.2011.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim HR, Kang HS, Kim HD. Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life. 1999;48(4):429–433. doi: 10.1080/713803536. [DOI] [PubMed] [Google Scholar]
- 18.McCollum AK, Lukasiewicz KB, Teneyck CJ, Lingle WL, Toft DO, Erlichman C. Cisplatin abrogates the geldanamycin-induced heat shock response. Mol Cancer Ther. 2008;7(10):3256–3264. doi: 10.1158/1535-7163.MCT-08-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. A pathway of multi-chaperone interactions common to diverse regulatory proteins: estrogen receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1, and the aryl hydrocarbon receptor. Cell Stress Chaperones. 1996;1(4):237–250. doi: 10.1379/1466-1268(1996)001<0237:apomci>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitesell L, Bagatell R, Falsey R. The stress response: implications for the clinical development of hsp90 inhibitors. Curr Cancer Drug Targets. 2003;3(5):349–358. doi: 10.2174/1568009033481787. [DOI] [PubMed] [Google Scholar]
- 21.Winklhofer KF, Reintjes A, Hoener MC, Voellmy R, Tatzelt J. Geldanamycin restores a defective heat shock response in vivo. J Biol Chem. 2001;276(48):45160–45167. doi: 10.1074/jbc.M104873200. [DOI] [PubMed] [Google Scholar]
- 22.Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ, Whitesell L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6(8):3312–3318. [PubMed] [Google Scholar]
- 23.Burris HA, 3rd, Berman D, Murthy B, Jones S. Tanespimycin pharmacokinetics: a randomized dose-escalation crossover phase 1 study of two formulations. Cancer Chemother Pharmacol. 2011;67(5):1045–1054. doi: 10.1007/s00280-010-1398-6. [DOI] [PubMed] [Google Scholar]
- 24.Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat Rev. 2013;39(4):375–387. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Goldman JW, Raju RN, Gordon GA, El-Hariry I, Teofilivici F, Vukovic VM, Bradley R, Karol MD, Chen Y, Guo W, Inoue T, Rosen LS. A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer. 2013;13:152. doi: 10.1186/1471-2407-13-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hong D, Said R, Falchook G, Naing A, Moulder S, Tsimberidou AM, Galluppi G, Dakappagari N, Storgard C, Kurzrock R, Rosen LS. Phase I study of BIIB028, a selective heat shock protein 90 inhibitor, in patients with refractory metastatic or locally advanced solid tumors. Clin Cancer Res. 2013;19(17):4824–4831. doi: 10.1158/1078-0432.CCR-13-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sessa C, Shapiro GI, Bhalla KN, Britten C, Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA, Akimov M, Quadt C, Fernandez-Ibarra C, Lu H, Bailey S, Chica S, Banerji U. First-inhuman phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(13):3671–3680. doi: 10.1158/1078-0432.CCR-12-3404. [DOI] [PubMed] [Google Scholar]