

Abstract
MHC molecule plays a key role in immunology, and the molecule binding reaction with peptide is an important prerequisite for T cell immunity induced. MHC II molecules do not have conserved residues, so they appear as open grooves. As a consequence, this will increase the difficulty in predicting MHC II molecules binding peptides. In this paper, we aim to propose a novel prediction method for MHC II molecules binding peptides. First, we calculate sequence similarity and structural similarity between different MHC II molecules. Then, we reorder pseudosequences according to descending similarity values and use a weight calculation formula to calculate new pocket profiles. Finally, we use three scoring functions to predict binding cores and evaluate the accuracy of prediction to judge performance of each scoring function. In the experiment, we set a parameter α in the weight formula. By changing α value, we can observe different performances of each scoring function. We compare our method with the best function to some popular prediction methods and ultimately find that our method outperforms them in identifying binding cores of HLA-DR molecules.
1. Introduction
Histocompatibility refers to the degree of antigenic similarity between the tissues of different individuals, which determines the acceptance or rejection of allografts. Transplantation antigen or histocompatibility antigen is the cause of rejection of allografts [1, 2]. MHC (Major Histocompatibility Complex) is present on the chromosome encoding a major histocompatibility antigen, mutual recognition between control cells, and the regulation of immune response.
MHC molecule plays a key role in immunology, and the molecule binding reaction with peptide is an important prerequisite for T cell immunity induced [2, 3]. By detecting a wide variety of microbial pathogens, the immune system protects host against diseases. Because of this, the binding prediction of MHC molecules with peptides has always been a hot topic in bioinformatics. Many researches in this field not only help us to understand the process of immune but also develop the work of vaccine design assisted by computers.
MHC genes produce two different types of molecules, which are MHC I molecules and MHC II molecules [1, 2]. MHC I molecules contain two separate polypeptide chains: the MHC α chain encoded by MHC genes and the MHC β chain encoded by non-MHC genes [4, 5]. MHC I class molecules are expressed in almost all eukaryotic cell surfaces, recognized by CD8+ cells. MHC II class molecules consist of two non-covalently linked polypeptide chains, namely, α chain and β chain. MHC II class molecules are expressed on antigen-presenting cells in general. Foreign MHC II antigens only capture and present on the surface of antigen-presenting cells (APC) TH cell [6]. After that, APC secretes large amounts of cytoplasm, activating cell invasion defensed behavior. Only the binding of antigen peptides and MHC II class molecules can activate CD4+ TH cells (helper T cells) [7]. Then, the activated TH cells would differentiate into effector cells and activate the immune response.
The structures of MHC I molecules and MHC II molecules slightly differ in the binding grooves [5]. Close grooves form on the binding of MHC I molecules and antigenic peptides. On the other hand, MHC II molecules do not have conserved residues, so they appear as open grooves. As a consequence, this will increase the difficulty in predicting MHC II molecules binding peptides [7]. In this paper, we aim to solve more difficult problem of predicting MHC II binding peptides.
The pioneering and most popular pan-specific approach for MHC II binding prediction is the TEPITOPE method [8], and basic idea is the HLA-DR allele having identical pseudosequence. The same pocket will share the same quantitative profile. By using multiple instance learning, the MHCIIMulti method [9] can predict more than 500 HLA-DR molecules. Transforming each DRB allele into a pseudosequence with 21 amino acids and using the SMM-align method to identify binding cores, the NetMHCIIpan method [5] gets an accurate prediction by using an artificial neural network algorithm [10, 11]. Combining NN-align and NetMHCpan with NetMHCIIpan [9, 12], the MULTIPRED2 method [13–15] can get a perfect prediction for 1077 HLA-I and HLA-II alleles and 26 HLA supertypes.
In this paper, we propose a novel prediction method for predicting MHC II molecules binding peptides. First, we calculate sequence similarity and structural similarity between different MHC molecules [13, 16]. Then, we reorder pseudosequences according to descending similarity values and use a weight calculation formula to calculate new pocket profiles. Finally, we use three scoring functions to predict binding cores and evaluate the accuracy of prediction to judge performance of each scoring function [17, 18]. In the experiments, we set a parameter α in the weight formula. By changing α value, we can observe different performances of each of the scoring functions. We compare our method with the best function to some popular prediction methods and ultimately find that our method outperforms them in identifying binding cores of HLA-DR molecule [19]. The work would suggest a novel computational strategy for special protein identification instead of traditional machine learning based methods [20, 21].
2. Materials and Methods
2.1. Data Sets
We find 39 MHC molecules and peptides binding complexes from Protein Data Bank (http://www.rcsb.org/pdb/search/), which constitutes the data set used in this paper. In this data set, lengths are between 11 and 23, and we can find polypeptide-binding sites, namely, binding cores. Table 1 lists the details of these 39 MHC molecules and peptide binding complexes [14, 22, 23].
Table 1.
Details of 39 MHC molecules and peptide binding complexes.
| PDB ID | DRB allele | Peptide sequence |
|---|---|---|
| 1AQD | DRB1∗0101 | VGSDWRFLRGYHQYA |
| 1PYW | DRB1∗0101 | XFVKQNAAALX |
| 1KLG | DRB1∗0101 | GELIGILNAAKVPAD |
| 1KLU | DRB1∗0101 | GELIGTLNAAKVPAD |
| 2FSE | DRB1∗0101 | AGFKGEQGPKGEPG |
| 1SJH | DRB1∗0101 | PEVIPMFSALSEG |
| 1SJE | DRB1∗0101 | PEVIPMFSALSEGATP |
| 1T5W | DRB1∗0101 | AAYSDQATPLLLSPR |
| 1T5X | DRB1∗0101 | AAYSDQATPLLLSPR |
| 2IAN | DRB1∗0101 | GELIGTLNAAKVPAD |
| 2IAM | DRB1∗0101 | GELIGILNAAKVPAD |
| 2IPK | DRB1∗0101 | XPKWVKQNTLKLAT |
| 1FYT | DRB1∗0101 | PKYVKQNTLKLAT |
| 1R5I | DRB1∗0101 | PKYVKQNTLKLAT |
| 1HXY | DRB1∗0101 | PKYVKQNTLKLAT |
| 1JWM | DRB1∗0101 | PKYVKQNTLKLAT |
| 1JWS | DRB1∗0101 | PKYVKQNTLKLAT |
| 1JWU | DRB1∗0101 | PKYVKQNTLKLAT |
| 1LO5 | DRB1∗0101 | PKYVKQNTLKLAT |
| 2ICW | DRB1∗0101 | PKYVKQNTLKLAT |
| 2OJE | DRB1∗0101 | PKYVKQNTLKLAT |
| 2G9H | DRB1∗0101 | PKYVKQNTLKLAT |
| 1A6A | DRB1∗0301 | PVSKMRMATPLLMQA |
| 1J8H | DRB1∗0401 | PKYVKQNTLKLAT |
| 2SEB | DRB1∗0401 | AYMRADAAAGGA |
| 1BX2 | DRB1∗1501 | ENPVVHFFKNIVTPR |
| 1YMM | DRB1∗1501 | ENPVVHFFKNIVTPRGGSGGGGG |
| 1FV1 | DRB5∗0101 | NPVVHFFKNIVTPRTPPPSQ |
| 1H15 | DRB5∗0101 | GGVYHFVKKHVHES |
| 1ZGL | DRB5∗0101 | VHFFKNIVTPRTPGG |
| 4E41 | DRB1∗0101 | GELIGILNAAKVPAD |
| 1DLH | DRB1∗0101 | PKYVKQNTLKLAT |
| 1KG0 | DRB1∗0101 | PKYVKQNTLKLAT |
| 3L6F | DRB1∗0101 | APPAYEKLSAEQSPP |
| 3PDO | DRB1∗0101 | KPVSKMRMATPLLMQALPM |
| 3PGD | DRB1∗0101 | KMRMATPLLMQALPM |
| 3S4S | DRB1∗0101 | PKYVKQNTLKLAT |
| 3S5L | DRB1∗0101 | PKYVKQNTLKLAT |
| 1HQR | DRB5∗0101 | VHFFKNIVTPRTP |
In Table 1, the first column is PDB ID of 39 complexes from PDB; the second column is the name of corresponding alleles from 39 complexes; the third column is the corresponding polypeptide sequences, in which the enlarged nine positions are the binding cores.
2.2. Methods
There are thousands of allele variants in nature [2, 4]. It is absolutely impossible to measure the binding specificity one by one. Motivated by this perspective, we propose a new computational method to predict the binding specificity of peptides without any biochemical experiment, which combines the sequence and structural information of these known specificity-binding MHC molecules, as showed in Figure 1. We evaluate the method on all general HLA-DRB data sets, and results indicate that our method is close to the state-of-the-art technology and our approach can predict all sequence-known MHC molecules and cost little time, extending the prediction space compared with other time-consuming approaches.
Figure 1.

The architecture of our approach to MHC II and peptide binding problem.
2.3. Crucial Pockets relative to Binding Specificities of HLA-DR Molecules
We mainly use Position Specific Scoring Matrix (PSSM) [13, 24] in our approach, which is a popular technology in the problem of MHC binding. Roughly speaking, there are nine amino acids in MHC binding cores, and each position is a specific pocket as showed in Table 2. We use PSSM to quantify the binding affinity between twenty basic amino acids with these nine pockets.
Table 2.
30 HLA-complexes binding pockets.
| PDB ID | Pocket 1 | Pocket 2 | Pocket 3 | Pocket 4 | Pocket 5 | Pocket 6 | Pocket 7 | Pocket 8 | Pocket 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1AQD | 82N 85V 86G | 77T 78Y 81H 82N | 78Y | 13F 74A 78Y | 13F 71R | 11L | 47Y 61W 67L 70Q 71R | 60Y 61W | 9W 57D 61W |
|
| |||||||||
| 1PYW | 82N 85V 86G 89F | 77T 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 13F 71R | 11L | 11L 28E 61W 71R | 60Y 61W | 57D 61W |
|
| |||||||||
| 1KLG | 82N 85V | 78Y 81H 82N | 78Y | 13F 71R 78Y | 13F 71R | 11L | 61W | 60Y 61W | 57D 61W |
|
| |||||||||
| 2FSE | 82N 85V 86G 89F | 77T 78Y 82N | 13F 28E 70Q 71R 74A 78Y | 13F 71R | 71R | 28E 47Y 61W 67L 71R |
61W | 57D | |
|
| |||||||||
| 1KLU | 82N 85V | 78Y 81H 82N | 13F 71R 78Y | 13F 71R | 11L | 61W | 60Y 61W | 57D 61W | |
|
| |||||||||
| 1SJH | 82N | 78Y 81H 82N | 13F 26L 70Q 71R 74A 78Y | 71R | 11L | 61W | 60Y 61W | 57D 61W | |
|
| |||||||||
| 1SJE | 82N | 78Y 81H 82N | 78Y | 13F 26L 70Q 71R 74A 78Y | 71R | 11L | 61W | 60Y 61W | 57D 60Y 61W |
|
| |||||||||
| 1T5W | 82N 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 13F 71R | 11L | 61W 71R | 60Y 61W | 9W 57D 61W |
|
| |||||||||
| 1T5X | 82N 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 71R | 11L | 61W 71R | 61W | 57D 61W |
|
| |||||||||
| 2IAN | 82N 85V | 78Y 81H 82N | 78Y | 13F 70Q 74A 78Y | 13F 70Q 71R | 11L | 61W 71R | 61W | 57D 61W |
|
| |||||||||
| 2IPK | 82N 85V 86G 89F | 77T 78Y 81H 82N | 13F 70Q 71R 74A 78Y | 71R | 11L | 47Y 61W 67L 71R | 60Y 61W | 9W 57D 61W | |
|
| |||||||||
| 1FYT | 82N 85V 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 13F 71R | 11L | 28E 47Y 61W 67L 71R | 60Y 61W | 9W 57D 61W |
|
| |||||||||
| 1R5I | 82N 85V 86G 89F | 77T 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 70Q 71R | 11L | 47Y 61W 67L 71R | 61W | 9W 57D 61W |
|
| |||||||||
| 1HXY | 82N 85V 86G 89F | 78Y 81H 82N | 13F 70Q 71R 74A 78Y | 71R | 11L | 28E 47Y 61W 67L 71R | 60Y 61W | 9W 57D 61W | |
|
| |||||||||
| 1JWM | 82N 85V 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 71R | 11L | 28E 47Y 61W 67L 71R | 61W | 57D 61W |
|
| |||||||||
| 1JWS | 82N 85V 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 13F 71R | 11L | 47Y 61W 67L 71R | 61W | 9W 57D 61W |
|
| |||||||||
| 1JWU | 82N 85V 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 13F 71R | 11L | 28E 47Y 61W 67L 71R | 61W | 9W 57D 61W |
|
| |||||||||
| 1LO5 | 82N 85V 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 78Y | 13F 71R | 11L | 47Y 61W 67L 71R | 61W | 9W 57D 60Y 61W |
|
| |||||||||
| 2ICW | 82N 85V 86G 89F | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 13F 71R | 11L | 28E 47Y 61W 67L 71R | 61W | 9W 57D 61W |
|
| |||||||||
| 2OJE | 82N 85V 86G | 77T 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 70Q 71R | 11L | 28E 47Y 61W 67L 71R | 61W | 9W 57D 61W |
|
| |||||||||
| 2G9H | 82N 85V 86G 89F | 77T 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 71R | 11L 13F | 28E 47Y 61W 67L 71R | 60Y 61W | 9W 57D 61W |
|
| |||||||||
| 2IAM | 82N | 78Y 81H 82N | 78Y | 13F 70Q 71R 74A 78Y | 70Q 71R | 11L | 61W 67L 71R |
60Y 61W | 57D 61W |
|
| |||||||||
| 1A6A | 82N 85V 86V | 77T 78Y 81H 82N | 78Y | 13S 26Y 74R 78Y | 71K 74R | 11S 30Y | 30Y 47F 61W 67L 71K | 61W | 9E 30Y 57D 61W |
|
| |||||||||
| 1J8H | 82N 85V 86G 89F | 77T 78Y 81H 82N | 78Y | 13H 26F 28D 70Q 74A 78Y |
13H 70Q 71K | 11V 13H 30Y | 30Y 47Y 61W 67L | 60Y 61W | 37Y 57D 61W |
|
| |||||||||
| 2SEB | 82N | 77T 78Y 81H 82N | 13H 26F 71K 78Y |
13H 71K | 30Y | 30Y 47Y 61W | 60Y 61W | 61W | |
|
| |||||||||
| 1BX2 | 82N 85V | 77T 78Y 81H 82N | 78Y | 13H 26F 28D 70Q 74A 78Y |
70Q | 13R | 57D 60Y 61W | ||
|
| |||||||||
| 1YMM | 82N | 77T 78Y 81H 82N | 78Y | 13R 26F 28D 70Q 74A 78Y |
70Q | 13R | 61W 67I | 61W | 57D 61W |
|
| |||||||||
| 1FV1 | 82N 85V 86G 89F | 78Y 81H 82N | 78Y | 13Y 71R 78Y | 71R | 13Y | 61W 67L 71K |
61W | 57D |
|
| |||||||||
| 1H15 | 82N 89F | 77T 78Y 81H 82N | 78Y | 13Y 71R 78Y | 71R | 11D 13Y 30D | 61W | 57D 60Y | |
|
| |||||||||
| 1ZGL | 82N 85V 89F | 77T 78Y 81H 82N | 13Y 26F 71R 78Y | 13Y | 13Y 28H 61W 71R | 61W | 57D 60Y 61W | ||
There are five anchor sites (1, 4, 6, 7, and 9) at the binding core for MHC II molecules, which determine the binding strength of peptides with MHC II molecules. Because site 1 of MHC II is consistent with different MHC II molecules and peptides, it is important to identify the precise quantification of its binding core in site 1, yet we use weights of four anchor sites (4, 6, 7, and 9) to define profiles. For other sites, the same approach, such as TEPITOPE, is to specify their quantitative profiles.
2.4. Computing Similarity between Different MHC Molecules
2.4.1. Sequence-Based Similarity
Sequence-based similarity can be calculated by alignment results. Here, pocket pseudosequences and associated profiles refer to raw pocket pseudosequences and raw pocket profiles, respectively. These raw pseudosequences are composed of several amino acids, whose associated residue indices are shown in Table 3. Eleven representative HLA-DR alleles are adopted to specify different profiles for anchor pockets 4, 6, 7, and 9. These eleven alleles are DRB1∗0101, DRB1∗0301, DRB1∗0401, DRB1∗0402, DRB1∗0404, DRB1∗0701, DRB1∗0801, DRB1∗1101, DRB1∗1302, DRB1∗1501, and DRB5∗0101. If two alleles have identical pseudosequences in the same pocket, they will have identical profiles. For a given pocket, we collect all the different raw pocket pseudosequences into one set R x, R x = {r 1, r 2,…, r m}, and |r i| = n, where i = 1,2,…, m, x ∈ {4, 6, 7, 9}, m is the number of unique pseudosequences, and n is the number of amino acids contained in a pseudosequence. Meanwhile, we collect all different raw profiles into one set P x, P x = {p 1, p 2,…, p m}, and |p i| = 20, where i = 1,2,…, m. There is a one-to-one correspondence between p i and r i. We use BLOSUM to calculate the sequence similarity between different MHC molecules, defined as BLOSUM = (S q − S i). Then, we can get encoded pseudosequence, which is a 20n-dimensional real vector V x = {V 1, V 2,…, V m}. We use Radial Basis Function (RBF) to measure the similarity between encoded predicted pseudosequences V a and a raw encoded pseudosequence:
| (1) |
Table 3.
Important positions at the binding core for MHC II molecules.
| Important positions | |
|---|---|
| Pocket 1 | 82 85 86 89 |
| Pocket 2 | 77 78 81 82 |
| Pocket 3 | 78 |
| Pocket 4 | 11 13 26 28 70 71 74 78 |
| Pocket 5 | 11 13 28 70 71 74 |
| Pocket 6 | 11 13 28 70 71 74 |
| Pocket 7 | 11 28 30 47 61 67 70 71 |
| Pocket 8 | 60 61 |
| Pocket 9 | 9 30 37 57 60 61 |
2.4.2. Structure-Based Similarity
Using MHC II HLA-peptide complex structure from Protein Data Bank (PDB), we can get the residues 3D-coordinate of the pocket in each MHC molecule, h (p x, p y, p z). We define vector H x = {h 1, h 2,…, h n}, where n is the number of amino acids in the pseudocontained sequence; meanwhile, we collect a set S x, S x = {H 1, H 2,…, H m}, m is the number of different pseudosequences, and there is also one-to-one correspondence between H i and r i.
Next, we need to estimate the similarity of three-dimensional structures between a measured MHC molecule and five MHC molecules with known pseudosequence PSSM. Rigid transformation is to compare three-dimensional substructures of two proteins [25, 26].
Intuitively, we fix one of the structures, A, move (translation and rotation) the other structure, B, and find the best movement in three-dimensional space, with two atoms to the nearest structure. We calculate the Euclidean distance between two structures, defined as RMSD = |C q − C i|. We can get encoded pseudosequence V x = {V 1, V 2,…, V m} and calculate the similarity between 3D structures of encoded predicted pseudosequences V a and a raw encoded pseudosequence:
| (2) |
2.4.3. Overall Similarity
After that, we have obtained sequence similarity and structural similarity. We calculate final similarity score functions according to the following three formulas:
| (3) |
2.5. Weights Calculation for New Pocket Profiles
We reorder all pseudosequences according to descending similarity values and use a weight calculation formula to calculate new pocket profiles. A new pocket profile is generated as a weighted average over m raw pocket profiles in P x. Next, we use the gamma distribution to generate the weights. The gamma PDF distribution is defined as follows:
| (4) |
where x > 0 and k, θ > 0, and γ(k) denotes the gamma function.
The weight distribution is generated to discretize the gamma PDF as follows:
| (5) |
where m is the dimension of the weights and k and θ are the shape and scale parameters, respectively. The gamma distribution generates the weight vector to give a higher weight for more similarity pseudosequences.
After normalizing, the weight vector is defined as follows:
| (6) |
Given a predicted DRB allele a, let K a = (K a1,K a2,…, K am), where K ai = K(V a, V i), V i ∈ V x, and α is a positive number and enhances the weight vector to protect the outstanding contribution of most similarity pseudosequences. Associated raw pocket profiles are P x = {P 1, P 2,…, P m}. Elements of K a are sorted in descending order, and the reordered vector of K a is denoted as . The corresponding weight vector is denoted as W = (ω 1, ω 2,…, ω m). We denote pocket profiles associated with the reordered vector as , . We define the pocket profile for allele a as follows:
| (7) |
where x ∈ {4,6, 7,9}.
3. Result
First, we design an experiment to choose appropriate scoring function to combine sequence similarity and structural similarity. Then, we compare with other state-of-the-art technologies, which are TEPITOPE, MultiRTA, NetMHCIIpan-2.0, and NetMHCIIpan-1.0. The result indicates that our approach can obtain better prediction and effectively extend current prediction methods. Finally, we test on more data sets.
3.1. Evaluation of Different Scoring Functions
Here, we use 30 of 39 MHC molecules and peptide complexes as test set and get the appropriate scoring functions as showed above. The value of the parameter α is set to 1, 2, 3, 4, 5, 10, 15, and 20, followed by results shown in Figure 2. We find that no significant changes can be found by K 1(V a, V i); for K 2(V a, V i) and K 3(V a, V i), when α = 1 prediction error number is 10 and 9 and when α = 3 prediction errors reduced to 8, we set the value of α to 3. Comparing these three functions, the least numbers of errors by three functions are 4, 8, and 8. Details are shown in Tables S1, S2, and S3, in the Supplementary Material available online at http://dx.doi.org/10.1155/2016/3832176.
Figure 2.
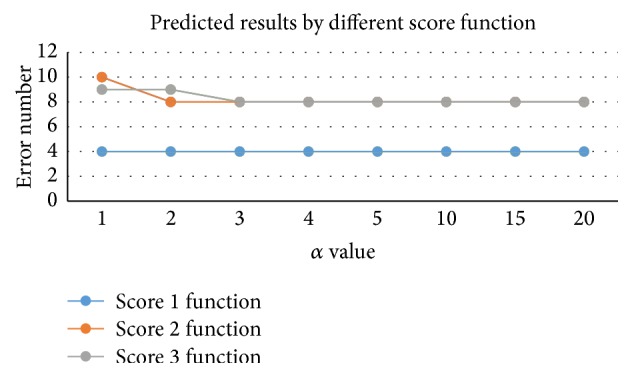
Predicted results by different score functions. x-axis represents different α values, and the y-axis refers to predicted results of different score functions.
3.2. Compared with Conventional Well-Known Methods
From the above experimental results, K 1(V a, V i) obtains the most accurate prediction, so we will select K 1(V a, V i) with α = 3 as our final approach. We compare our current prediction results with conventional well-known methods TEPITOPE [23], MultiRTA [13], NetMHCIIpan-2.0 [12], and NetMHCIIpan-1.0 [12], and these results are shown in Table 4.
Table 4.
Comparison of our binding prediction with other approaches. The 5th column is the result of our method, and 6th to 8th columns are results of TEPITOPE, MultiRTA, and NetMHCIIpan. The bold cell means one error.
| PDB ID | Allele | Peptide | Core | Ours | TEPITOPE | MultiRTA | NetMHCIIpan-2.0 |
|---|---|---|---|---|---|---|---|
| 1AQD | DRB1∗0101 | VGSDWRFLRGYHQYA | WRFLRGYHQ | WRFLRGYHQ | WRFLRGYHQ | WRFLRGYHQ | WRFLRGYHQ |
| 1PYW | DRB1∗0101 | XFVKQNAAALX | FVKQNAAAL | FVKQNAAAL | FVKQNAAAL | FVKQNAAAL | FVKQNAAAL |
| 1KLG | DRB1∗0101 | GELIGILNAAKVPAD | IGILNAAKV | IGILNAAKV | IGILNAAKV | IGILNAAKV | LIGILNAAK |
| 2FSE | DRB1∗0101 | GELIGTLNAAKVPAD | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV |
| 1KLU | DRB1∗0101 | AGFKGEQGPKGEPG | FKGEQGPKG | FKGEQGPKG | FKGEQGPKG | FKGEQGPKG | FKGEQGPKG |
| 1SJH | DRB1∗0101 | PEVIPMFSALSEG | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS |
| 1SJE | DRB1∗0101 | PEVIPMFSALSEGATP | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS | VIPMFSALS |
| 1T5W | DRB1∗0101 | AAYSDQATPLLLSPR | YSDQATPLL | SDQATPLLL | YSDQATPLL | SDQATPLLL | YSDQATPLL |
| 1T5X | DRB1∗0101 | AAYSDQATPLLLSPR | YSDQATPLL | SDQATPLLL | YSDQATPLL | SDQATPLLL | YSDQATPLL |
| 2IAN | DRB1∗0101 | GELIGTLNAAKVPAD | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV | IGTLNAAKV |
| 2IPK | DRB1∗0101 | GELIGILNAAKVPAD | IGILNAAKV | IGILNAAKV | IGILNAAKV | IGILNAAKV | LIGILNAAK |
| 1FYT | DRB1∗0101 | XPKWVKQNTLKLAT | WVKQNTLKL | WVKQNTLKL | WVKQNTLKL | WVKQNTLKL | WVKQNTLKL |
| 1R5I | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 1HXY | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 1JWM | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 1JWS | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 1JWU | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 1LO5 | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 2ICW | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 2OJE | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 2G9H | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 2IAM | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 1A6A | DRB1∗0301 | PVSKMRMATPLLMQA | MRMATPLLM | MRMATPLLM | MRMATPLLM | MRMATPLLM | MRMATPLLM |
| 1J8H | DRB1∗0401 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL | YVKQNTLKL |
| 2SEB | DRB1∗0401 | AYMRADAAAGGA | MRADAAAGG | MRADAAAGG | MRADAAAGG | MRADAAAGG | YMRADAAAG |
| 1BX2 | DRB1∗1501 | ENPVVHFFKNIVTPR | VHFFKNIVT | VHFFKNIVT | VHFFKNIVT | VHFFKNIVT | VVHFFKNIV |
| 1YMM | DRB1∗1501 | ENPVVHFFKNIVTPRGGSGGGGG | VHFFKNIVT | VHFFKNIVT | VHFFKNIVT | VHFFKNIVT | VHFFKNIVT |
| 1FV1 | DRB5∗0101 | NPVVHFFKNIVTPRTPPPSQ | FKNIVTPRT | KNIVTPRTP | FKNIVTPRT | VHFFKNIVT | FFKNIVTPR |
| 1H15 | DRB5∗0101 | GGVYHFVKKHVHES | YHFVKKHVH | YHFVKKHVH | YHFVKKHVH | YHFVKKHVH | YHFVKKHVH |
| 1ZGL | DRB5∗0101 | VHFFKNIVTPRTPGG | FKNIVTPRT | KNIVTPRTP | FKNIVTPRT | VHFFKNIVT | FFKNIVTPR |
|
| |||||||
| Results | 4 errors | 0 errors | 4 errors | 6 errors | |||
TEPITOPE is a relatively early method and is one of the most popular methods for predicting MHC II binding molecules. The basic idea is that if two HLA-DR alleles have the same pseudorandom sequence in the same pocket, they share the same number of profiles. Through multiple instances, MHCIIMulti has predicted over 500 HLA-DR molecules. NetMHCIIpan firstly converts each of the DRB alleles into a pseudorandom sequence of 21 amino acids, then uses the SMM-align method to identify binding residues in the peptide chain and the core side, and finally uses artificial neural network to train the model. MultiRTA makes prediction on HLA-DR and HLA-DP molecules. By thermodynamic method, it calculates a peptide chain and all other residues to predict the average binding affinity of binding strength and the introduction of standardization constraints to avoid overfitting. MULTIPRED2 can predict 1077 HLA-I and HLA-II genes and 26 HLA supertypes. Details are as shown in Figure 3. Our method obtains 4 errors; however, TEPITOPE, MultiRTA, NetMHCIIpan-2.0, and NetMHCIIpan-1.0 get the numbers of errors as 0, 4, 6, and 3, respectively. Because now we only find five MHC II molecules with three-dimensional structural information, we use the scoring matrix with only 5 MHC II molecules. If the three-dimensional structural information of MHC II molecules can be extended to all of the 11 MHC II molecules, our predictions will be more accurate. From the current view, our approach has reached a higher level of prediction.
Figure 3.

Comparison of different methods by sequence logos of peptides on HLA-DRB1∗0101.
3.3. Other Prediction Results
When compared with other methods on the above experiments, we only use 30 of 39 MHC molecules and peptide complexes as test set. In this section, we test on the remaining nine MHC molecules. In this experiment, we choose K 1(V a, V i) and set the parameter α = 3. As seen in Table 5, eight of nine predictions are accurate. Therefore, our approach produces a considerably great performance.
Table 5.
Other prediction results of nine MHC molecules. This table shows the prediction result of our method on 9 MHC molecules. The 5th column is the result. There is only one error result, which is shown using bold font.
| PDB ID | Allele | Peptide | Core | Ours |
|---|---|---|---|---|
| 4E41 | DRB1∗0101 | GELIGILNAAKVPAD | IGILNAAKV | IGILNAAKV |
| 1DLH | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL |
| 1KG0 | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL |
| 3L6F | DRB1∗0101 | APPAYEKLSAEQSPP | YEKLSAEQS | YEKLSAEQS |
| 3PDO | DRB1∗0101 | KPVSKMRMATPLLMQALPM | MRMATPLLM | KMRMATPLL |
| 3PGD | DRB1∗0101 | KMRMATPLLMQALPM | MRMATPLLM | MRMATPLLM |
| 3S4S | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL |
| 3S5L | DRB1∗0101 | PKYVKQNTLKLAT | YVKQNTLKL | YVKQNTLKL |
| 1HQR | DRB5∗0101 | VHFFKNIVTPRTP | FKNIVTPRT | FKNIVTPRT |
|
| ||||
| Results | 1 error | |||
4. Conclusion
In this paper, we try to solve the problem of predicting MHC II binding peptides with a novel metric and strategy. Sequence similarity and structural similarity between different MHC molecules are calculated to reorder pseudosequences according to descending similarity, and then a weight calculation formula is used to calculate new pocket profiles. Finally, we use three scoring functions to predict binding cores and evaluate the accuracy of prediction to judge performance of each scoring function. In the experiment, we set a parameter α in the weight formula. By changing α value, we can observe different performances of each scoring function. Then, we compare our method with the best function to some popular prediction methods and ultimately find that our method outperforms them in identifying binding cores of HLA-DR molecules.
Supplementary Material
Using different functions to combine sequence similarity and structural similarity, these are the predicted results with the value of alpha ranging from 1 to 5.
Acknowledgments
This work is supported by a grant from the National Science Foundation of China (NSFC 61402326) and Peiyang Scholar Program of Tianjin University (no. 2016XRG-0009).
Competing Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Zinkernagel R. M., Doherty P. C. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248(5450):701–702. doi: 10.1038/248701a0. [DOI] [PubMed] [Google Scholar]
- 2.Terasaki P. I. A brief history of HLA. Immunologic research. 2007;38(1–3):139–148. doi: 10.1007/s12026-007-0020-4. [DOI] [PubMed] [Google Scholar]
- 3.Maenaka K., Jones E. Y. MHC superfamily structure and the immune system. Current Opinion in Structural Biology. 1999;9(6):745–753. doi: 10.1016/S0959-440X(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 4.Robinson J., Waller M. J., Fail S. C., et al. The IMGT/HLA database. Nucleic Acids Research. 2009;37(supplement 1):D1013–D1017. doi: 10.1093/nar/gkn662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen M., Lundegaard C., Blicher T., et al. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Computational Biology. 2008;4(7, article e1000107) doi: 10.1371/journal.pcbi.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rammensee H.-G., Bachmann J., Emmerich N. P. N., Bachor O. A., Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3):213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 7.Germain R. N. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76(2):287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 8.Sturniolo T., Bono E., Ding J., et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nature Biotechnology. 1999;17(6):555–561. doi: 10.1038/9858. [DOI] [PubMed] [Google Scholar]
- 9.Pfeifer N., Kohlbacher O. Multiple instance learning allows MHC class II epitope predictions across alleles. Algorithms in Bioinformatics. 2008;5251:210–221. [Google Scholar]
- 10.Kindt T. J., Goldsby R. A., Osborne B. A., Kuby J. Kuby Immunology. New York, NY, USA: WH Freeman & Company; 2007. [Google Scholar]
- 11.Sette A., Adorini L., Appella E., et al. Structural requirements for the interaction between peptide antigens and I-Ed molecules. Journal of Immunology. 1989;143(10):3289–3294. [PubMed] [Google Scholar]
- 12.Nielsen M., Lundegaard C., Blicher T., et al. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Computational Biology. 2008;4(7) doi: 10.1371/journal.pcbi.1000107.e1000107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bordner A. J., Mittelmann H. D. MultiRTA: a simple yet reliable method for predicting peptide binding affinities for multiple class II MHC allotypes. BMC Bioinformatics. 2010;11, article 482 doi: 10.1186/1471-2105-11-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reche P. A., Zhang H., Glutting J.-P., Reinherz E. L. EPIMHC: a curated database of MHC-binding peptides for customized computational vaccinology. Bioinformatics. 2005;21(9):2140–2141. doi: 10.1093/bioinformatics/bti269. [DOI] [PubMed] [Google Scholar]
- 15.Reche P. A., Glutting J.-P., Zhang H., Reinherz E. L. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics. 2004;56(6):405–419. doi: 10.1007/s00251-004-0709-7. [DOI] [PubMed] [Google Scholar]
- 16.Sette A., Adorini L., Appella E., et al. Structural requirements for the interaction between peptide antigens and I-Ed molecules. The Journal of Immunology. 1989;143(10):3289–3294. [PubMed] [Google Scholar]
- 17.Nielsen M., Justesen S., Lund O., Lundegaard C., Buus S. NetMHCIIpan-2.0—improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Research. 2010;6(1, article 9) doi: 10.1186/1745-7580-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen M., Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10, article 296 doi: 10.1186/1471-2105-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang G. L., DeLuca D. S., Keskin D. B., et al. MULTIPRED2: a computational system for large-scale identification of peptides predicted to bind to HLA supertypes and alleles. Journal of Immunological Methods. 2011;374(1-2):53–61. doi: 10.1016/j.jim.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng X.-Y., Huang W.-J., Hu S.-C., et al. A global characterization and identification of multifunctional enzymes. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0038979.e38979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou Q., Wang Z., Guan X., Liu B., Wu Y., Lin Z. An approach for identifying cytokines based on a novel ensemble classifier. BioMed Research International. 2013;2013:11. doi: 10.1155/2013/686090.686090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen W., Zhang S., Wong H. An effective and effecient peptide binding prediction approach for a broad set of HLA-DR molecules based on ordered weighted averaging of binding pocket profiles. Proteome Science. 2013;11:p. S15. doi: 10.1186/1477-5956-11-s1-s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L., Chen Y., Wong H.-S., Zhou S., Mamitsuka H., Zhu S. TEPITOPEpan: extending TEPITOPE for peptide binding prediction covering over 700 HLA-DR molecules. PLoS ONE. 2012;7(2) doi: 10.1371/journal.pone.0030483.e30483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reche P. A., Reinherz E. L. Prediction of peptide-MHC binding using profiles. Methods in Molecular Biology. 2007;409:185–200. doi: 10.1007/978-1-60327-118-9_13. [DOI] [PubMed] [Google Scholar]
- 25.Guo F., Li S. C., Wang L., Zhu D. Protein-protein binding site identification by enumerating the configurations. BMC Bioinformatics. 2012;13, article 158 doi: 10.1186/1471-2105-13-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo F., Li S. C., Wang L. Protein-protein binding sites prediction by 3D structural similarities. Journal of Chemical Information and Modeling. 2011;51(12):3287–3294. doi: 10.1021/ci200206n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Using different functions to combine sequence similarity and structural similarity, these are the predicted results with the value of alpha ranging from 1 to 5.