

Abstract
Amide bond-forming reactions are of tremendous significance in synthetic chemistry. Methodological research has, in the past, focused on efficiency and selectivity, and these have reached impressive levels. However, the unacceptable amounts of waste produced have led the ACS GCI Roundtable to label ‘amide bond formation avoiding poor atom economy' as the most pressing target for sustainable synthetic method development. In response to this acute demand, we herein disclose an efficient one-pot amide coupling protocol that is based on simple alkynes as coupling reagents: in the presence of a dichloro[(2,6,10-dodecatriene)-1,12-diyl]ruthenium catalyst, carboxylate salts of primary or secondary amines react with acetylene or ethoxyacetylene to vinyl ester intermediates, which undergo aminolysis to give the corresponding amides along only with volatile acetaldehyde or ethyl acetate, respectively. The new amide synthesis is broadly applicable to the synthesis of structurally diverse amides, including dipeptides.
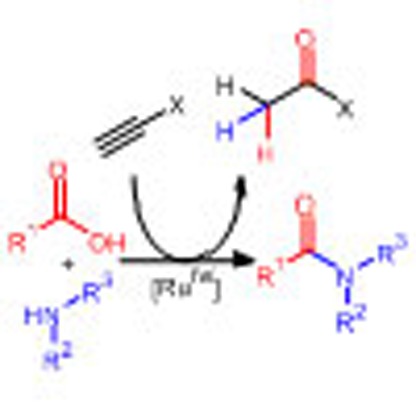 Amide formation is a ubiquitous reaction in organic chemistry, but suffers from the problem of generating large amounts of waste. Here, the authors report a catalytic amide synthesis by the ruthenium catalysed addition of carboxylic acids to acetylenes, followed by reaction with primary or secondary amines.
Amide formation is a ubiquitous reaction in organic chemistry, but suffers from the problem of generating large amounts of waste. Here, the authors report a catalytic amide synthesis by the ruthenium catalysed addition of carboxylic acids to acetylenes, followed by reaction with primary or secondary amines.
Amide bond formation is one of the most frequently used transformations in organic chemistry1,2,3,4. The most desirable amide synthesis, a direct condensation of carboxylic acids with amines, is hindered by the intrinsic acid–base reactivity of the starting materials. The thermal amide bond formation from the ammonium carboxylate salts requires high temperatures5,6,7, which can be lowered by Lewis acids or boronic acid derivatives. However, even the best known systems are limited to a narrow range of amines and require scavenging the reaction water, for example, by large amounts of molecular sieves. (Fig. 1, left)8,9,10,11,12,13. Therefore, amides are usually synthesized by aminolysis of activated carboxylic acid derivatives, such as halides, anhydrides, azides, or activated esters, that are mostly generated in an extra step with aggressive, expensive or waste-intensive reagents14,15,16,17,18,19,20. The other main strategy for amide bond formation involves the in situ activation of carboxylic acids by peptide coupling reagents, such as carbodiimides or phosphonium salts21,22,23,24,25,26,27,28,29,30,31. Such amide syntheses are highly optimized and provide access to almost any amide structure in near quantitative yields. In modern protein synthesis, they are complemented by efficient chemical and enzymatic peptide ligation methods32,33,34,35,36,37. However, the atom economy of all these processes is low, and the cumulative waste generated during amide synthesis is unacceptable. As a result, the ACS GCI Roundtable has identified ‘amide bond formation avoiding poor atom economy' as the most pressing target for sustainable synthetic method development38.
Figure 1. Atom-efficient approaches to amide bond formation.

(a) Thermal or Lewis acid-mediated dehydration of ammonium carboxylates. (b) Catalytic addition of alcohols to nitriles. (c) Dehydrogenative coupling of alcohols with amines. (d) Oxidative coupling of aldehydes and amines. (e) Oxidative coupling of alkynes and amines.
Over the last years, some elegant strategies for waste-minimized amide synthesis have been devised (Fig. 1), for example, dehydrogenative couplings of alcohols, aldehydes or alkynes with amines, or additions of alcohols to nitriles39,40,41,42,43,44,45,46,47,48,49,50,51. However, for most synthetic organic chemists, carboxylic acids and amines are still the optimal substrate base for amide synthesis.
To address the central issue of atom economy in the synthesis of amides from ammonium carboxylates, we looked for an activator with minimal molecular weight and low intrinsic reactivity that would scavenge the reaction water in a catalytic condensation process. We envisioned that a hydroacyloxylation catalyst with unprecedented activity might enable the generation of vinyl esters from ammonium carboxylates and gaseous acetylene. Aminolysis of these intermediates would furnish the desired amides along with volatile acetaldehyde.
RuII, AgI and AuI complexes efficiently promote the addition of carboxylic acids to alkynes under mild conditions, as reported by Mitsudo, Dixneuf, Bruneau and others52,53,54,55,56,57,58,59. The aminolysis of enol esters takes place under similarly mild conditions60,61,62,63. However, for all known catalysts, the two reaction steps are incompatible. As a result, this technology appeared limited to two-step procedures with isolation of sensitive enol esters. For example, Kita et al. reported an amide synthesis via isolated ketene acetal intermediates64, and Breinbauer et al. synthesized polypeptides via a Ru-catalysed hydroacyloxylation of alkynes followed by enzymatic aminolysis65. These reactions demonstrate the potential of this concept, giving access to amides in high yields under mild conditions, as demanded especially by peptide chemists. However, this approach can reach synthetic maturity only through a catalytic one-pot process that overcomes all its associated problems, for example, carboxylate salt formation with basic amines which hinders catalytic hydroacyloxylation, the control of hydroamination as a side reaction, and the challenging activation of gaseous acetylene, which state-of-the-art catalysts have not been extending to66.
We disclose herein an amidation protocol which allows the use of low-molecular acetylene and its more activated homologue ethoxyacetylene as a sustainable alternative for state-of-the-art coupling agents. These procedures are convincing in terms of the amount, toxicity and separation of the formed byproducts, yet, broadly applicable, convenient and comparable cheap.
Results
Development of a one-pot amide synthesis
Evaluation of state-of-the-art catalysts, for example, [Ru(methallyl)2dppb] or [RuCl2(PPh3)(p-cymene)]58,67,68,69, in the reaction between benzoic acid (1a) and 1-hexyne confirmed that they give high yields only in the absence of benzylamine. None of them catalysed the reaction of 1a with acetylene to give vinyl benzoate (3a; Supplementary Tables 4 and 5).
However, we were pleased to find that simple RuCl3 catalyses the conversion of benzylammonium benzoate (6aa) to the desired N-benzyl benzamide in up to 75% yield at 80 °C under acetylene at 1.7 bar, which is its usual tank pressure (Table 1, entry 1). Systematic evaluation of RuIII and RuIV precursors revealed that Ru-1 was most effective (entries 2 and 3). Phosphine and nitrogen ligands adversely affected the yield (Supplementary Tables 1 and 2). This is surprising, because the only known RuIV hydroacyloxylation catalyst is the triphenylphosphine complex reported by Cadierno et al.70
Table 1. One-pot activation and amidation of carboxylic acids with acetylene*.

Dioxane was found to be the best solvent, but the reaction also works well in toluene, THF and ethyl acetate (entries 4−8). The reaction is surprisingly tolerant to oxygen and water up to a certain threshold (Supplementary Table 1).
Under optimal conditions, that is, 2 mol% Ru-1 or RuCl3 in dioxane at 80 °C, the amide forms in near quantitative yield within 6 h with acetylene as the carboxylate activator (Table 1, entry 9, Supplementary Table 1). Higher alkynes are inactive as activators, but with ethoxyacetylene and Ru-1 as catalyst, full conversion was observed already at 40 °C within 4 h (entries 13−15). Under identical conditions, RuCl3 gives only unsatisfactory yields for this activator (Supplementary Table 2). The advantages of the somewhat less atom-economic ethoxyacetylene are that it is more easily handled on small scales than gaseous acetylene, and that inert ethyl acetate rather than acetaldehyde is released.
Both new protocols were compared with two-step procedures using established catalysts64, in which the enol esters are formed in a separate step, with consecutive addition of the amine either in the same solvent or after solvent exchange. With acetylene as the activator, no conversion could be achieved, and with ethoxyacetylene, the yields obtained in these two-step syntheses were much lower than those obtained with our convenient one-step protocols (Supplementary Tables 1 and 2).
Applicability of the developed processes
The scope of the ecologically and economically beneficial acetylene protocol is illustrated in Table 2. Aliphatic, aromatic and heteroaromatic carboxylates were successfully coupled with primary amines. Unfortunately, the substrate scope of this protocol is limited by the solubility of the alkylammonium carboxylates in dioxane, the optimal solvent for acetylene gas.
Table 2. Scope of the amidation with acetylene as the activating agent*.

Such restrictions do not apply to the ethoxyacetylene protocol in the solvent N-methyl-2-pyrrolidone, which is applicable to a remarkably wide range of substrates (Table 3). Aromatic, heteroaromatic and aliphatic carboxylic acids reacted with benzylamine to give high yields of the corresponding amides. Diverse functionalities including halo, ether, amide, aldehyde, ester and even-free OH groups were tolerated. Other primary and secondary amines were successfully converted to the corresponding benzamides in good to excellent yields when increasing the temperature to 80 °C to ensure full conversion (Supplementary Table 3). Remarkably, the coupling of less nucleophilic compounds such as amides, aniline and diethylamine with benzoic acid also gave the desired products, albeit in low yields. Other oxygen- or sulfur-based nucleophiles could not be converted.
Table 3. Scope of the amidation with ethoxyacetylene as activating agent*.

The synthetic concept may also be used for peptide couplings. Various N-protected amino acids were successfully coupled with amino acid esters. Without additives, racemization could not fully be suppressed but remained below 10%, which is a good basis for dedicated optimization.
Mechanistic considerations
The reaction mechanism was investigated by in situ nuclear magnetic resonance spectroscopy. The experiments confirmed the intermediacy of enol esters, which formed within minutes and were consumed in the course of the reaction (see Supplementary Table 6 and Supplementary Fig. 1 respectively). We thus conclude that as outlined in Fig. 2, the reaction proceeds via a Ru-catalysed hydroacyloxylation via a standard catalytic cycle67,71 followed by aminolysis. In ESI MS investigations of the reaction mixture, species with m/z values of 754 and 647 were dominant. These were identified as [RuCl2(benzyl amine)3(ethoxyacetylene)2(benzoate)]+ and [RuCl2(benzyl amine)2(ethoxyacetylene)2(benzoate)]+. In tandem mass spectrometry (MS) experiments, these adducts fragmented with loss of benzyl amine ligands and formation of the six-coordinate [RuCl2(benzyl amine)1(ethoxyacetylene)2(benzoate)]+ complex, which we believe to be the catalyst resting state. It is reasonable to assume that it is a Ru(IV)-complex, since it bears three anions and is still positively charged. These investigations suggest the intermediacy of high-valent Ru-species, which explains why RuIV pecursors have a higher activity than the RuII and Ru0 precursors employed in other catalytic additions. For the details of the spectroscopic investigation, see Supplementary Figs 2–5. In-depth, studies are required to clarify whether the carboxylate addition proceeds via Ru-complexes with η2-coordinated alkynes or via Ru-alkylidene complexes.
Figure 2. Catalytic amide condensation via enol esters.

The proposed catalytic cycle starts with the coordination of a carboxylate and an alkyne to the ruthenium catalyst, followed by an addition of the carboxylate to the alkyne. After protonolysis, the enol ester intermediate is released, which then acts as an acylating agent for the amine, yielding the desired amide along with the carbonyl-byproduct.
In conclusion, the feasibility of catalytic amidation reactions with minimal waste production has been demonstrated. Even though extensive optimization is still required, this reaction concept could become an important factor in meeting one of the key challenges of Green Chemistry.
Methods
For analytical data and preparation methods of the compounds in this article, see Supplementary Figs 6–111 and Supplementary Methods.
General techniques
All reactions were performed in oven-dried glassware containing a Teflon-coated stirring bar and dry septum under a nitrogen atmosphere. For the exclusion of atmospheric oxygen from the reaction media, solvents were degassed by argon sparge and purified by standard procedures before use. Non-aqueous amines were distilled before use. All reactions were monitored by gas chromatography (GC) using n-tetradecane as an internal standard or by high-performance liquid chromatography using anisole as an internal standard. Response factors of the products with regard to n-tetradecane/anisole were obtained experimentally by analysing known quantities of the substances. GC analyses were carried out using an HP-5 capillary column (Phenyl Methyl Siloxane 30 m × 320 × 0.25, 100/2.3-30-300/3) and a temperature programme beginning with 2 min at 60 °C followed by 30 °C/min ramp to 300 °C, then 3 min at this temp. High-performance liquid chromatography analyses were carried out using a Shimadzu LC-2010A. The stationary phase was a reversed phase column LiChroCart PAH C-18 from Merck KGaA with acetonitrile and water as eluents at 60 °C and the following solvent programme: starting from 10 vol% acetonitrile for 1 min, followed by increasing acetonitrile to 70 vol% during 23 min, then decreasing again to 10 vol% rapidly and maintaining this value for the next 2 min. Column chromatography was performed using a Combi Flash Companion-Chromatography-System (Isco-Systems) and RediSep packed columns (12 g). nuclear magnetic resonance spectra were obtained on Bruker AMX 400 or on Bruker Avance 600 systems using DMSO-d6, Chloroform-d3 or Toluene-d8 as solvent, with proton and carbon resonances at 400/600 MHz and 101/151 MHz, respectively. Mass spectral data were acquired on a GC-MS Saturn 2,100 T (Varian). Infrared spectra were recorded on Perkin Elmer Spectrum 100 FT-IR Spectrometer with Universal ATR Sampling Accessory. Melting points are uncorrected and were measured on a Mettler FP 61. ESI MS data were acquired on a Bruker Esquire 6,000 and evaluated with mMass software. Sample solutions at concentrations of ∼1 × 10−4 M were continuously infused into the ESI chamber at a flow rate of 2 μl min−1 using a syringe pump. We use nitrogen as drying gas at a flow rate of 3.0–4.0 l min−1 at 300 °C and spray the solutions at a nebulizer pressure of 4 psi with the electrospray needle held at 4.5 kV. CHN-elemental analyses were performed with a Hanau Elemental Analyzer vario Micro cube and HRMS with a Waters GCT Premier.
Synthesis of amides using acetylene as activator
An oven-dried headspace vial with Teflon-coated stirring bar was charged with the corresponding ammonium carboxylate (1 mmol) and dichloro[(2,6,10-dodecatriene)-1,12-diyl]ruthenium (5.06 mg, 20 μmol). The atmosphere was changed three times with nitrogen, then N-methylpyrrolidone (1 ml) and the corresponding amine (0.5 mmol) were added. The vial was placed in an autoclave reactor, the atmosphere was changed twice with acetylene, and a pressure of 1.7 bar was set. The mixture was then heated to 80 °C for 6 h. After cooling down to room temperature, the mixture was diluted with 20 ml of ethyl acetate and washed with each 20 ml of saturated NaHCO3 solution, water and brine. The organic layer was dried with MgSO4, the solvent removed under reduced pressure and the residue purified by column chromatography (SiOH, ethyl acetate/cyclohexane gradient).
Synthesis of amides using ethoxyacetylene as activator
An oven-dried headspace vial with Teflon-coated stirring bar was charged with the corresponding carboxylic acid (1 mmol) and dichloro[(2,6,10-dodecatriene)-1,12-diyl]ruthenium (5.06 mg, 15.0 μmol). The atmosphere was changed three times with nitrogen, then N-methylpyrrolidone (2 ml), benzyl amine (164 mg, 167 μl, 1.5 mmol) and ethoxyacetylene (40 wt%-solution in hexane; 210 mg, 299 μl and 1.5 mmol) were added in this order. The mixture was then heated to 40 °C for 4 h. After cooling down to room temperature, the mixture was diluted with 20 ml of ethyl acetate and washed with each 20 ml of sat. NaHCO3 solution, water and brine. The organic layer was dried with MgSO4, the solvent removed under reduced pressure and the residue purified by column chromatography (SiOH, ethyl acetate/cyclohexane gradient).
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files.
Additional information
How to cite this article: Krause, T. et al. Atom-economic catalytic amide synthesis from amines and carboxylic acids activated in situ with acetylenes. Nat. Commun. 7:11732 doi: 10.1038/ncomms11732 (2016).
Supplementary Material
Supplementary Figures 1-111, Supplementary Tables 1-6, Supplementary Methods and Supplementary References
Acknowledgments
We thank Astra Zeneca, DFG (SFB/TRR-88, ‘3MET') and Deutsche Bundesstiftung Umwelt (fellowship to S.B.) for financial support, Umicore AG for the donation of chemicals and Johannes Lang for technical assistance performing ESI MS measurements.
Footnotes
Author contributions L.J.G. planned and supervised the research; T.K. conceived and performed most experiments together with S.B.; B.E. performed additional experiments; T.K. and S.B. isolated and characterized the products; T.K. and L.J.G. co-wrote the manuscript.
References
- Montalbetti C. A. G. N. & Falque V. Amide bond formation and peptide coupling. Tetrahedron 61, 10827–10852 (2005). [Google Scholar]
- Lanigan R. M. & Sheppard T. D. Recent developments in amide synthesis: direct amidation of carboxylic acids and transamidation reactions. Eur. J. Org. Chem. 2013, 7453–7465 (2013). [Google Scholar]
- Pattabiraman V. R. & Bode J. W. Rethinking amide bond synthesis. Nature 480, 471–479 (2011). [DOI] [PubMed] [Google Scholar]
- Lundberg H., Tinnis F., Selander N. & Adolfsson H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 43, 2714–2742 (2014). [DOI] [PubMed] [Google Scholar]
- Gooßen L. J., Ohlmann D. M. & Lange P. P. The thermal amidation of carboxylic acids revisited. Synthesis 2009, 160–164 (2009). [Google Scholar]
- Mitchell J. A. & Reid E. E. The preparation of aliphatic amides. J. Am. Chem. Soc. 53, 1879–1883 (1931). [Google Scholar]
- Allen C. L., Chhatwal A. R. & Williams J. M. J. Direct amide formation from unactivated carboxylic acids and amines. Chem. Commun. 48, 666–668 (2012). [DOI] [PubMed] [Google Scholar]
- Nelson P. & Pelter A. 954. Trisdialkylaminoboranes: new reagents for the synthesis of enamines and amides. J. Chem. Soc. 5142–5144 (1965). [Google Scholar]
- Ishihara K., Ohara S. & Yamamoto H. 3, 4, 5-Trifluorobenzeneboronic acid as an extremely active amidation catalyst. J. Org. Chem. 61, 4196–4197 (1996). [DOI] [PubMed] [Google Scholar]
- Tinnis F., Lundberg H. & Adolfsson H. Direct catalytic formation of primary and tertiary amides from non-activated carboxylic acids, employing carbamates as amine source. Adv. Synth. Catal. 354, 2531–2536 (2012). [Google Scholar]
- Allen C. L. & Williams J. M. J. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 40, 3405–3415 (2011). [DOI] [PubMed] [Google Scholar]
- Mohy El Dine T., Erb W., Berhault Y., Rouden J. & Blanchet J. Catalytic chemical amide synthesis at room temperature: One more step toward peptide synthesis. J. Org. Chem. 80, 4532–4544 (2015). [DOI] [PubMed] [Google Scholar]
- Lundberg H. & Adolfsson H. Hafnium-Catalyzed direct amide formation at room temperature. ACS Catal. 5, 3271–3277 (2015). [Google Scholar]
- Villeneuve G. B. & Chan T. H. A rapid, mild and acid-free procedure for the preparation of acyl chlorides including formyl chloride. Tetrahedron Lett. 38, 6489–6492 (1997). [Google Scholar]
- Lal G. S., Pez G. P., Pesaresi R. J., Prozonic F. M. & Cheng H. Bis(2-methoxyethyl)aminosulfur Trifluoride: a new broad-spectrum deoxofluorinating agent with enhanced thermal stability. J. Org. Chem. 64, 7048–7054 (1999). [Google Scholar]
- Shioiri T., Ninomiya K. & Yamada S. Diphenylphosphoryl azide. new convenient reagent for a modified Curtius reaction and for peptide synthesis. J. Am. Chem. Soc. 94, 6203–6205 (1972). [DOI] [PubMed] [Google Scholar]
- Carpino L. A., Beyermann M., Wenschuh H. & Bienert M. Peptide synthesis via amino acid halides. Acc. Chem. Res. 29, 268–274 (1996). [Google Scholar]
- Lee J. B. Preparation of acyl halides under very mild conditions. J. Am. Chem. Soc. 88, 3440–3441 (1966). [Google Scholar]
- Olah G. A., Nojima M. & Kerekes I. Synthetic methods and reactions; IV. 1 Fluorination of carboxylic acids with cyanuric fluoride. Synthesis 1973, 487–488 (1973). [Google Scholar]
- Carpino L. A. & El-Faham A. Tetramethylfluoroformamidinium hexafluorophosphate: a rapid-acting peptide coupling reagent for solution and solid phase peptide synthesis. J. Am. Chem. Soc. 117, 5401–5402 (1995). [Google Scholar]
- Windridge G. & Jorgensen E. C. 1-Hydroxybenzotriazole as a racemization-suppressing reagent for the incorporation of im-benzyl-L-histidine into peptides. J. Am. Chem. Soc. 93, 6318–6319 (1971). [DOI] [PubMed] [Google Scholar]
- Kisfaludy L., Schőn I., Szirtes T., Nyéki O. & Lőw M. A novel and rapid peptide synthesis. Tetrahedron Lett. 15, 1785–1786 (1974). [Google Scholar]
- König W. & Geiger R. Eine neue methode zur synthese von peptiden: aktivierung der carboxylgruppe mit dicyclohexylcarbodiimid unter zusatz von 1-hydroxy-benzotriazolen. Chem. Ber. 103, 788–798 (1970). [DOI] [PubMed] [Google Scholar]
- Kisfaludy L. & Schön I. Preparation and applications of pentafluorophenyl esters of 9-fluorenylmethyloxycarbonyl amino acids for peptide synthesis. Synthesis 1983, 325–327 (1983). [Google Scholar]
- Mikozlajczyk M. & Kiezlbasiński P. Recent developments in the carbodiimide chemistry. Tetrahedron 37, 233–284 (1981). [Google Scholar]
- Carpino L. A. & El-Faham A. The diisopropylcarbodiimide/ 1-hydroxy-7-azabenzotriazole system: segment coupling and stepwise peptide assembly. Tetrahedron 55, 6813–6830 (1999). [Google Scholar]
- Guinó M. & Kuok (Mimi) H. K. Wang-aldehyde resin as a recyclable support for the synthesis of α,α-disubstituted amino acid derivatives. Org. Biomol. Chem. 3, 3188–3193 (2005). [DOI] [PubMed] [Google Scholar]
- Coste J., Frérot E., Jouin P. & Castro B. Oxybenzotriazole free peptide coupling reagents for N-methylated amino acids. Tetrahedron Lett. 32, 1967–1970 (1991). [Google Scholar]
- Han S.-Y. & Kim Y.-A. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron 60, 2447–2467 (2004). [Google Scholar]
- Valeur E. & Bradley M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 38, 606–631 (2009). [DOI] [PubMed] [Google Scholar]
- Gabriel C. M., Keener M., Gallou F. & Lipshutz B. H. Amide and peptide bond formation in water at room temperature. Org. Lett. 17, 3968–3971 (2015). [DOI] [PubMed] [Google Scholar]
- Dawson P., Muir T., Clark-Lewis I. & Kent S. Synthesis of proteins by native chemicalligation. Science 266, 776–779 (1994). [DOI] [PubMed] [Google Scholar]
- Bode J. W., Fox R. M. & Baucom K. D. Chemoselective amide ligations by decarboxylative condensations of N-alkylhydroxylamines and α-ketoacids. Angew. Chem. Int. Ed. 45, 1248–1252 (2006). [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xu C., Lam H. Y., Lee C. L. & Li X. Protein chemical synthesis by serine and threonine ligation. Proc. Natl Acad. Sci. USA 110, 6657–6662 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson B. L., Kiessling L. L. & Raines R. T. Staudinger ligation: a peptide from a thioester and azide. Org. Lett. 2, 1939–1941 (2000). [DOI] [PubMed] [Google Scholar]
- Noda H., Erős G. & Bode J. W. Rapid ligations with equimolar reactants in water with the potassium acyltrifluoroborate amide formation. J. Am. Chem. Soc. 136, 5611–5614 (2014). [DOI] [PubMed] [Google Scholar]
- Fouché M., Masse F. & Roth H.-J. Hydroxymethyl salicylaldehyde auxiliary for a glycine-dependent amide-forming ligation. Org. Lett. 17, 4936–4939 (2015). [DOI] [PubMed] [Google Scholar]
- Constable D. J. C. et al. Key green chemistry research areas: a perspective from pharmaceutical manufacturers. Green Chem. 9, 411–420 (2007). [Google Scholar]
- Tamaru Y., Yamada Y. & Yoshida Z. Direct oxidative transformation of aldehydes to amides by palladium catalysis. Synthesis 1983, 474–476 (1983). [Google Scholar]
- Tillack A., Rudloff I. & Beller M. Catalytic amination of aldehydes to amides. Eur. J. Org. Chem. 2001, 523–528 (2001). [Google Scholar]
- Yoo W.-J. & Li C.-J. Highly efficient oxidative amidation of aldehydes with amine hydrochloride salts. J. Am. Chem. Soc. 128, 13064–13065 (2006). [DOI] [PubMed] [Google Scholar]
- Gunanathan C., Ben-David Y. & Milstein D. Direct synthesis of amides from alcohols and amines with liberation of H2. Science 317, 790–792 (2007). [DOI] [PubMed] [Google Scholar]
- Ekoue-Kovi K. & Wolf C. One-pot oxidative esterification and amidation of aldehydes. Chem. Eur. J. 14, 6302–6315 (2008). [DOI] [PubMed] [Google Scholar]
- Dobereiner G. E. & Crabtree R. H. Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem. Rev. 110, 681–703 (2010). [DOI] [PubMed] [Google Scholar]
- De Sarkar S. & Studer A. Oxidative amidation and azidation of aldehydes by NHC catalysis. Org. Lett. 12, 1992–1995 (2010). [DOI] [PubMed] [Google Scholar]
- Chen C. & Hong S. H. Oxidative amide synthesis directly from alcohols with amines. Org. Biomol. Chem. 9, 20–26 (2011). [DOI] [PubMed] [Google Scholar]
- Zhang L. et al. Aerobic oxidative coupling of alcohols and amines over Au–Pd/resin in water: Au/Pd molar ratios switch the reaction pathways to amides or imines. Green Chem. 15, 2680–2684 (2013). [Google Scholar]
- Kang B., Fu Z. & Hong S. H. Ruthenium-catalyzed redox-neutral and single-step amide synthesis from alcohol and nitrile with complete atom economy. J. Am. Chem. Soc. 135, 11704–11707 (2013). [DOI] [PubMed] [Google Scholar]
- Li F., Ma J., Lu L., Bao X. & Tang W. Combination of gold and iridium catalysts for the synthesis of N-alkylated amides from nitriles and alcohols. Catal. Sci. Technol. 5, 1953–1960 (2015). [Google Scholar]
- Miyamura H., Min H., Soulé J.-F. & Kobayashi S. Size of gold nanoparticles driving selective amide synthesis through aerobic condensation of aldehydes and amines. Angew. Chem. Int. Ed. 54, 7564–7567 (2015). [DOI] [PubMed] [Google Scholar]
- Owston N. A., Parker A. J. & Williams J. M. J. Iridium-catalyzed conversion of alcohols into amides via oximes. Org. Lett. 9, 73–75 (2007). [DOI] [PubMed] [Google Scholar]
- Bruneau C. in Hydrofunctionalization eds Ananikov V. P., Tanaka M. 43, 203–230Springer Berlin Heidelberg (2011). [Google Scholar]
- Ishino Y., Nishiguchi I., Nakao S. & Hirashima T. Novel synthesis of enol esters through silver-catalyzed reaction of acetylenic compounds with carboxylic acids. Chem. Lett. 5, 641–644 (1981). [Google Scholar]
- Chary B. C. & Kim S. Gold(I)-catalyzed addition of carboxylic acids to alkynes. J. Org. Chem. 75, 7928–7931 (2010). [DOI] [PubMed] [Google Scholar]
- Rotem M. & Shvo Y. Addition of carboxylic acids to alkynes catalyzed by ruthenium complexes. Vinyl ester formation. Organometallics 2, 1689–1691 (1983). [Google Scholar]
- Mitsudo T., Hori Y. & Watanabe Y. Selective addition of unsaturated carboxylic acids to terminal acetylenes catalyzed by bis(.eta.5-cyclooctadienyl)ruthenium(II)-tri-n-butylphosphine. A novel synthesis of enol esters. J. Org. Chem. 50, 1566–1568 (1985). [Google Scholar]
- Ruppin C., Lecolier S. & Dixneuf P. H. Regioselective synthesis of isopropenyl esters by ruthenium catalysed addition of N-protected amino-acids to propyne. Tetrahedron Lett. 29, 5365–5368 (1988). [Google Scholar]
- Bruneau C., Neveux M., Kabouche Z., Ruppin C. & Dixneuf P. H. Ruthenium-catalysed additions to alkynes: synthesis of activated esters and their use in acylation reactions. Synlett 1991, 755–763 (1991). [Google Scholar]
- Gooßen L. J., Paetzold J. & Koley D. Regiocontrolled ru-catalyzed addition of carboxylic acids to alkynes: practical protocols for the synthesis of vinyl esters. Chem. Commun. 706–707 (2003). [PubMed] [Google Scholar]
- Kita Y. et al. Facile and efficient syntheses of carboxylic anhydrides and amides using (trimethylsilyl)ethoxyacetylene. J. Org. Chem. 51, 4150–4158 (1986). [Google Scholar]
- Kabouche Z., Bruneau C. & Dixneuf P. H. Enol esters as intermediates for the facile conversion of amino acids into amides and dipeptides. Tetrahedron Lett. 32, 5359–5362 (1991). [Google Scholar]
- Neveux M., Bruneau C., Lécolier S. & Dixneuf P. H. Novel syntheses of oxamides, oxamates and oxalates from diisopropenyl oxalate. Tetrahedron 49, 2629–2640 (1993). [Google Scholar]
- Bruneau C. & Dixneuf P. H. Selective transformations of alkynes with ruthenium catalysts. Chem. Commun. 6, 507–512 (1997). [Google Scholar]
- Kita Y., Maeda H., Omori K., Okuno T. & Tamura Y. Novel efficient synthesis of 1-ethoxyvinyl esters using ruthenium catalysts and their use in acylation of amines and alcohols: synthesis of hydrophilic 3′-N-acylated oxaunomycin derivatives. J. Chem. Soc. Perkin Trans. 1, 2999–3005 (1993). [Google Scholar]
- Schröder H. et al. Racemization-free chemoenzymatic peptide synthesis enabled by the ruthenium-catalyzed synthesis of peptide enol esters via alkyne-addition and subsequent conversion using alcalase-cross-linked enzyme aggregates. Adv. Synth. Catal. 355, 1799–1807 (2013). [Google Scholar]
- Ashton Acton Q. Benzoic Acids—Advances in Research and Application Scholarly Editions (2013). [Google Scholar]
- Doucet H., Martin-Vaca B., Bruneau C. & Dixneuf P. H. General synthesis of (Z)-alk-1-en-1-yl esters via ruthenium-catalyzed anti-Markovnikov trans-addition of carboxylic acids to terminal alkynes. J. Org. Chem. 60, 7247–7255 (1995). [Google Scholar]
- Doucet H., Höfer J., Bruneau C. & Dixneuf P. H. Stereoselective synthesis of Z-enol esters catalysed by [bis(diphenylphosphino)alkane]bis(2-methylpropenyl)ruthenium complexes. J. Chem. Soc. Chem. Commun. 850, 850–851 (1993). [Google Scholar]
- Gooßen L. J., Salih K. S. M. & Blanchot M. Synthesis of secondary enamides by ruthenium-catalyzed selective addition of amides to terminal alkynes. Angew. Chem. Int. Ed. 47, 8492–8495 (2008). [DOI] [PubMed] [Google Scholar]
- Cadierno V., Francos J. & Gimeno J. Ruthenium(IV)-catalyzed Markovnikov addition of carboxylic acids to terminal alkynes in aqueous medium. Organometallics 30, 852–862 (2011). [Google Scholar]
- Alonso F., Beletskaya I. P. & Yus M. Transition-metal-catalyzed addition of heteroatom−hydrogen bonds to alkynes. Chem. Rev. 104, 3079–3160 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-111, Supplementary Tables 1-6, Supplementary Methods and Supplementary References
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files.