

Abstract
We present two siblings with a partial deletion of chromosome 1p31.1 involving only the neuronal growth regulator 1 (NEGR1) gene. The siblings had a history of neuropsychiatric and behavioral problems, learning difficulties, hypotonia, mild aortic root dilatation, hypermobility, and scoliosis. This is the first clinical report of a microdeletion of chromosome 1p31.1 involving only the NEGR1 gene.
Keywords: partial chromosome 1p31.1 deletion, microarray, NEGR1 gene, visual performance, siblings, obesity, neuropsychiatry
Introduction
Interstitial deletions of chromosome 1p31 have been reported in at least 10 individuals and phenotypic characteristics summarized by Mircher et al.1 Clinical findings included intellectual disability, hypertonia, failure to thrive, microcephaly, broad nasal tip, tendency for an open mouth, micrognathia, short neck, fifth finger clinodactyly, and tapering fingers. Additional features described more recently included short stature, obesity, facial asymmetry, hypodontia,2 ventriculomegaly, hypogenesis of the corpus callosum, abnormal external genitalia,3 prominent supraorbital ridges, pectus excavatum, high-arched palate,4 strabismus, midline cleft of the upper and lower lip, and autism spectrum disorder5 6 (Table 1). Neuropsychiatric or behavioral phenotypes for chromosome 1p31 deletion have been poorly characterized to date beyond autism spectrum disorder and intellectual disability.5
Table 1. Clinical features of previously reported individuals with interstitial chromosome 1p31 deletionsa .
| Clinical features | Sibling 1 | Sibling 2 | Previously reported | Total |
|---|---|---|---|---|
| Intellectual disability | ± | ± | 8/12 | 10/14 |
| Autism spectrum disorder | ± | 1/1 | 2/3 | |
| Dyslexia | + | 1/2 | ||
| Short stature | + | 5/8 | 6/9 | |
| Obesity/overweight | 2/9 | 2/11 | ||
| Hypertonia | 3/6 | 3/8 | ||
| Microcephaly | 4/8 | 4/10 | ||
| Cleft lip/palate | 4/12 | 4/14 | ||
| Tendency for open mouth | 4/7 | 4/9 | ||
| Micrognathia | 6/5 | 6/7 | ||
| Short neck | 4/8 | 4/10 | ||
| Pectus deformity | 2/8 | 2/10 | ||
| Aortic root dilatation | + | + | 0/1 | 2/3 |
| Scoliosis | + | + | 4/6 | 6/8 |
| Joint laxity | + | + | 3/6 | 5/8 |
| Hypothyroidism | + | 1/2 | ||
| Projectile vomiting | + | 1/2 |
We present two siblings with neuropsychiatric and behavioral problems with an interstitial microdeletion of chromosome 1p31.1 involving only the neuronal growth regulator 1 (NEGR1) gene and provide clinical information about the partial deletion of the NEGR1 gene and its potential role in our siblings' phenotype. None of the previously reported cases showed involvement of only the NEGR1 gene (Fig. 1). This gene encodes NEGR1, which is associated with neuronal growth, proliferation, and differentiation7 and may contribute to body weight control and obesity.8
Fig. 1.
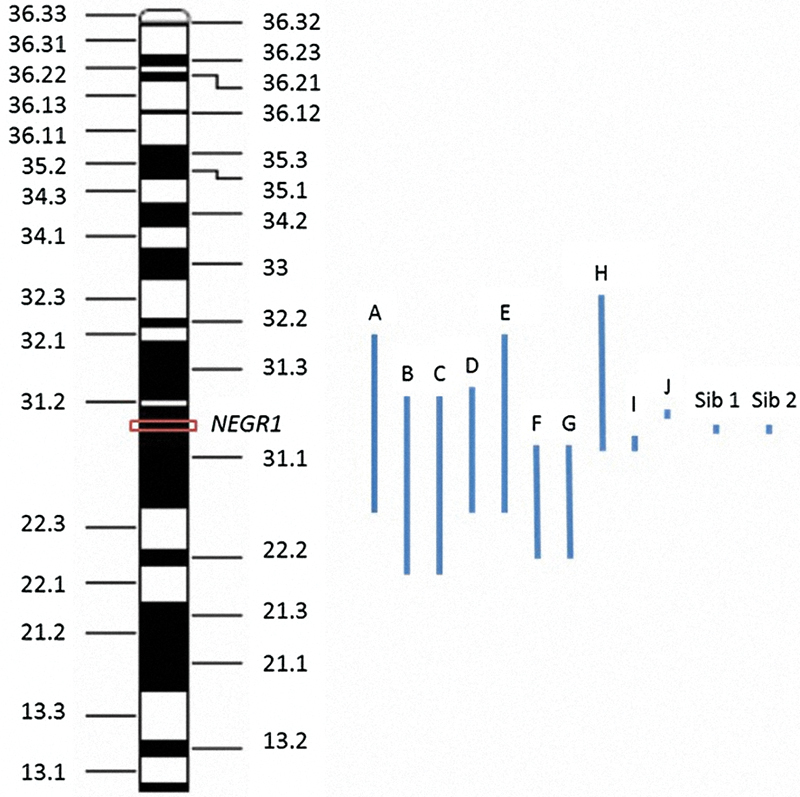
Location of interstitial chromosome 1p deletions for sibling 1, sibling 2, and others reported in the literature. The NEGR1 gene is indicated by a rectangular box on chromosome 1p31.1 (71,868,625–72,748,533bp, hg 19). Deletions of previously reported patients are indicated as follows: (A) del(1)(p22p32),7 (B) del(1)(p22.1p31.2),8 (C) patient 1 del(1)(p22.1p31.2),9 (D) patient 2 del(1)(p22.3p31.3),9 (E) del(1)(p22p32),1 (F) del(1)(p22.2p31.1),2 (G) del(1)(p22.2p31.1),4 (H) del(1)(p32p31),3 (I) del(1)(p31.1p31.1) (71,187,451–71,230,750)(hg 19),5 (J) del(1)(p31p31)(75,247,714–75,520,877, hg 19),6 sib 1 del(1)(p31.1p31.1)(71,656,103–72,077,213, hg19), and sib 2 del(1)(p31.1p31.1) (71,760,013–72,075,351, hg19).
Clinical Reports
Siblings 1 and 2 are African-American and Caucasian full siblings (one male and one female) who were adopted by the same family at less than 3 years of age. Their Caucasian biological mother is currently of 38 years. By report, she had a mild intellectual disability (severity unknown), history of bipolar disorder, and substance abuse during most of her adult life. Her height was 163 cm (40th centile) and weight was 72.6 kg (85th centile). Recreational drug and alcohol use was highly suspected during the pregnancies with both siblings; amount and type of drug use were unknown. The siblings have three maternal half-sisters who are 20, 15, and 8 years of age with two half-sisters having bipolar disorder but no further maternal family history available. Their biological father is African-American and suspected to have substance abuse. By report, his height was 173 cm (25th centile) and weight was 72.6 kg (55th centile). The father was unavailable; therefore, no additional paternal family history could be obtained.
Sibling 1
Sibling 1 was born at an estimated gestational age of 39 weeks via vaginal vertex delivery. His birth weight was 3,490 g (45th centile), Apgar scores are unknown. He was diagnosed with duplicated ureters at the age of 10 days, had corrective surgery, and was lost to follow-up. His developmental milestones were not documented. At 13 months of age, he had a near drowning event and a history of emotional and behavioral problems requiring medical intervention since early childhood. These problems included marked hyperactivity, impulsivity, persistent anxiety, aggressive behavior, recurrent tantrums, and early developmental delays in speech, social skills, communicative competence, and sensory issues. He had chronic insomnia. At the age of 6 years, his vision was 60/20, he wore an eye patch and now has a vision of 20/25 with corrective glasses. His reading skills are at a 2nd grade level, but math skills are described as an area of relative strength. Psychiatric diagnoses included attention deficit hyperactivity disorder (ADHD), sensory integration dysfunction, and speech articulation difficulty. Although he had features of autism, an assessment by developmental pediatrics including the autism diagnostic observation schedule was not diagnostic for an autism spectrum disorder. Other medical evaluations showed chronic kidney disease (duplicated ureters), scoliosis of 14 degrees and mild aortic root dilatation (Z score unavailable) and features resembling a connective tissue disorder.
He was evaluated at the age of 7 years at another facility because of the concerns regarding learning impairment. He was described as generally nondysmorphic although cupped-shaped ears, small epicanthal folds, and relatively narrow palpebral fissures were noted. The physical examination was negative for hearing problems, with an absence of birthmarks or skin conditions. His vision was recorded as 20/25 with eyeglasses. His height was 130 cm (90th centile), weight was 25.6 kg (75th centile), and his head circumference was 52.3 cm (50th centile). Chronic constipation and fatigue were also noted. Neurological examination was normal except for decreased tone and ligamentous laxity.
Sibling 1 was first seen by child psychiatry in our facility at the age of 8 years because of poor impulse control, irritability, and impulsive aggression. He was initially described as having cycles of irritability and crying, that occurred every 3 weeks. He was reading at the first grade level but could not write in sentences. Comprehension was better with spoken versus written language. At the age of 8 years 11 months, his full-scale intelligence quotient (IQ) using the Stanford Binet Intelligence Scale 5th edition was 86 (low-average range), with verbal IQ score of 97 (average) and nonverbal IQ score of 77 (borderline). Fluid reasoning score was 106 (average), working memory 83 (low average), knowledge 86 (low average), and quantitative reasoning 89 (average) and visual spatial processing 79 (borderline). Achievement testing measured with the Wechsler individual Achievement test 3rd edition showed his oral language composite score was 86 (average), in comparison to below average performance in reading. Total reading standard score was 73 (with standard scores in basic reading 75, reading comprehension and fluency 72, and written expression 73), while his receptive vocabulary, essay composition, and oral expression were average.
At the age of 9 years, sibling 1 underwent a visual performance profile because of the concerns raised about dyslexia or reading ability, specifically that he would transpose letters upon writing. He had a visual performance profile performed, which consisted of two parts, neuromuscular visual skills and visual thinking skills. For neuromuscular visual skills, he showed a normal range for smooth eye and saccadic eye control. He had significant deficiencies for only eye teaming. He had severe deficiencies with performance at the 7-year level in left-to-right tracking. For visual thinking skills, he had significant deficiencies with performance at the 7-year level for reversal and sentence copy tests. He also had severe deficiencies in visual discrimination (performance at 5 years 11 months), visualization (performance at < 5 years), visual memory (performance at < 5 years), visual form constancy (performance at 5 years 7 months), and visual closure (performance at 4 years 10 months). His testing indicated deficiencies in both neuromuscular visual skills and visual thinking skills, both of which are necessary for school success. A general binocular dysfunction was noted along with oculomotor dysfunction/pursuits and specific delays in development pertaining to visual processing. Ocular motor dysfunction deficiencies of pursuit eye movements are characterized by difficult visually tracking objects, loss of place or omission of words in lines of print while reading and transposition when copying from one source document to another.
During his most recent physical examination at the age of 11 years 9 months, he was a cooperative pleasant male with a head circumference of 52.7 cm (45th centile), inner canthal distance of 2.7 cm (23rd centile), outer canthal distance of 7.7 cm (15th centile), interpupillary distance of 5.5 cm (50th centile), palpebral fissure length of 2.7 cm (50th centile), and ear length of 6.2 cm (60th centile) (Fig. 2). His ears were prominent with poor appearing architecture and cartilage, epicanthal folds, enamel dysplasia, mild shawl scrotum, total hand length of 15.2 cm (20th centile), middle finger length of 6.2 cm (15th centile), mild 2 to 3 toe syndactyly bilaterally, shortened 5th phalanges, wide space between first and second toes bilaterally, hypopigmented skin patches on face, loose skin, and a Beighton evaluation score for Ehlers Danlos syndrome was 6 (of 9) for thumb hyperextension bilaterally, elbow hyperextension unilaterally, hyperextended knees bilaterally, and able to touch palms of hands to floor. His height measures ranged between the 20th and 40th percentiles over his lifespan. His height at 11 years 9 months was 141.5 cm (24th centile). His weight measures during the past had fallen to the 20th centile from measures previously at or around the 50th centile for most of his life. No change in his diet, exercise, or health status was noted. No large increase was noted in his height. During our physical examination at 11 years 9 months of age, his weight was 35.47 kg (25th centile).
Fig. 2.
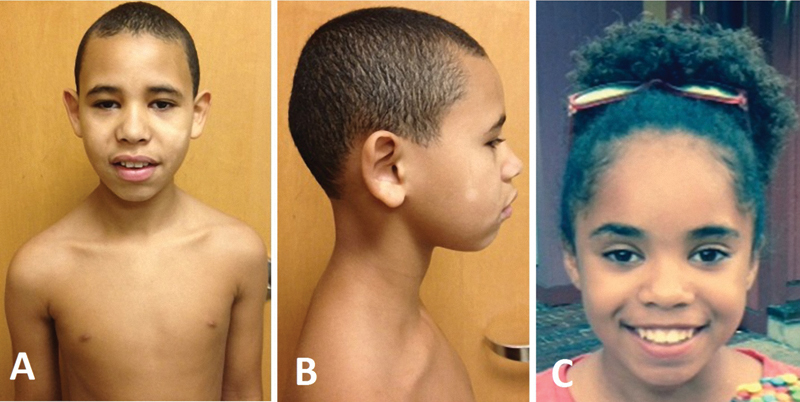
(A) Frontal and (B) profile views of 11-year-old male sibling 1 and (C) frontal view of a 12-year-old female sibling 2 both of whom have the 1p31.1 microdeletion involving only the NEGR1 gene.
Sibling 2
Sibling 2 is a 12-year-old female who is the older sister of sibling 1. She was referred to our Genetics clinic at the age of 11 years for developmental regression, ADHD, and family history of 1p31.1 microdeletion. She weighed 3,150 g (35th centile) at birth but had difficulty gaining weight throughout her life in spite of an adequate appetite with consuming a variety of food sources. Her growth parameters have been below average. There were no developmental delays noted at the time of her adoption at the age of 3 years. She was also found to have dilation of the aortic root (Z score of 2.2) as seen in her brother (sibling 1) and mitral valve prolapse by echocardiogram. She had a mild S-shaped scoliosis of the thoracolumbar spine and was given a clinical diagnosis of Ehlers–Danlos syndrome, hypermobility type by previous clinical evaluation. Sleep disturbances were noted and unexplained projectile vomiting reportedly occurs approximately once every 2 months.
Sibling 2 also received speech therapy services throughout elementary school and has a history of learning impairment. She developed behavioral problems associated with ADHD, memory deficits and recent developmental regression. At the age of 11 years, she had a Wechsler intelligent scale performed, which showed she had verbal and perceptual scores of 96, difficulties in working memory with a score of 88, and a processing speed of 85. Her full scale IQ score was 89. They noted that ADHD may have been the reason for her processing difficulties. She had a Vineland adaptive behavior examination, which showed a communication score of 81, a daily living score of 86, and a socialization score of 82 with a composite score of 77. Her reading skills were not assessed. No formal memory testing was performed.
At the time of evaluation, acquired primary hypothyroidism was noted along with delayed bone age (chronological age of 11 years, bone age of 9 years). She began treatment for her thyroid at the age of 11 years and growth hormone therapy for growth failure and retardation at the age of 12 years. Sibling 2 was 138 cm (3rd centile) in height and 30.6 kg (3rd centile) in weight. Her inner canthal distance was 2.7 cm (20th centile), outer canthal distance was 7.9 cm (20th centile), interpupillary distance was 5.6 cm (65th centile), palpebral fissure length was 2.5 cm (15th centile), and ear length of 5.2 cm (10th centile) (Fig. 2). Epicanthal folds, a protruding left ear, hirsutism, soft skin (but not hyperextensible), keloids, and hypermobile joints were noted. Genetic testing for connective tissue disorders was not performed, as she was lost to follow-up.
Genetic Testing
Genetic testing for fragile X syndrome for sibling 1 was negative (33 CGG repeats). He also had a chromosomal microarray analysis using approximately 180,000 copy number probes (Agilent Technologies, Santa Clara, California, United States) in a Clinical Laboratory Improvement Amendments approved commercial laboratory (Children's Mercy Hospital, Kansas City, Missouri, United States). A 421 kb deletion of chromosome 1p31.1 [71,656,103–72,077,213, GRCH 37, hg19] was found for sibling 1, which included three of the seven exons of the NEGR1 gene. Quantitative polymerase chain reaction was used to verify the presence of the deletion. No other microarray abnormalities were noted in the genome for sibling 1. Parental testing was unavailable.
Because of the history of her biological sibling having the chromosomal 1p31.1 microdeletion, sibling 2 had a 400 K comparative genomic hybridization—array performed including 300,000 copy number probes and 100,000 single-nucleotide polymorphism probes (Agilent Technologies) at a median probe spacing of 13 kb except for the X chromosome with probe spacing at 5 kb. The hybridization was done in a Clinical Laboratory Improvement Amendments approved commercial laboratory (Ambry Genetics, Irvine, California, United States). A partial deletion (315 kb in size) of the chromosomal 1p31.1 was found with coordinates chr1:71.760.013–72.075.351 (GRCh37 hg 19), a dozen probes were deleted within the chromosome 1p31.1 region (Fig. 3). No other microarray abnormalities were noted. Parental testing was unavailable.
Fig. 3.
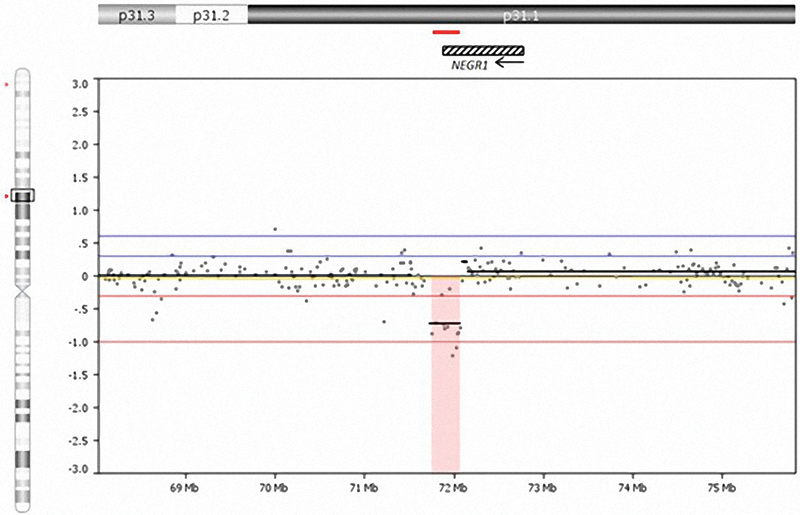
Interstitial deletion of chromosome 1p31.1 seen in sibling 2 showing the size and location of the deletion and position of the NEGR1 gene, which is partially deleted on chromosome 1. Arrow indicates direction of transcription of the NEGR1 gene, which consists of 7 exons positioned from right to left in transcription. The small horizontal line noted above the NEGR1 gene illustration represents the deletion size and location for sibling 2.
Discussion
Our male and female siblings both showed partial chromosome 1p31.1 deletions including only the NEGR1 gene. The array utilized for sibling 2 consisted of 400,000 probes covering the entire genome showing a dozen probes deleted within the chromosome 1p31.1 region. The deletion size seen in sibling 1 using a different microarray with fewer probes was estimated at a larger size. Quantitative polymerase chain reaction confirmed the presence of the 1p31.1 deletion. The smaller deletion size of 315 kb seen in sibling 2 may be more accurate because of the higher number of probes used in the newer, more dense microarray. The microdeletion seen in the siblings is unique and not reported in the previous subjects described in the literature involving chromosome 1p311 9 or in genomic databases.10 As the siblings have been adopted and parents' DNA was not available for further investigations, the inheritance of the copy number variation (CNV) could not be established. However, as the CNV was identified in the two siblings, it is unlikely to be de novo.
The NEGR1 gene, with genomic coordinates of chr1:71.868.625–72.748.533 (GRCH37, hg19), plays a role in neuronal growth, proliferation, and differentiation.11 The cloned rat NEGR1 gene is expressed in the neuronal processes of the adult rat cerebral cortex and hippocampus.11 Expression studies in mice found that NEGR1 is strongly expressed in the cerebral cortex, hippocampus, olfactory bulb, and hypothalamus.12 The NEGR1 gene encoded protein is distributed on dendrites and the soma of pyramidal neurons similar to LAMP (limbic system associated membrane protein), a cell recognition molecule.11 The NEGR1 gene, also termed Kilon, is proposed to play a role in obesity or body weight control12 with reported loss of function in knockout mouse models with abolished gene expression causing a reduction in body mass without a change in feeding habits with a standard chow diet. Up to 8% body mass reduction was seen in the males with the knockout genes and up to 13% body mass reduction in the knockout females.12 However, other studies involving both increased and decreased expression of the NEGR1 gene generated via adeno-associated virus models indicated that decreased expression of NEGR1 led to increases in chow intake resulting in increased body weight.13 Hence, the exact process and how NEGR1 plays a role in obesity remains to be discovered; but, it appears to be involved in locomotor activity and body temperature regulation.12 13 Sibling 2 was small in size and underweight but maintained a normal diet. These phenotypic features partially recapitulate what was observed in the knockout mouse model reported in the literature.12 Sibling 1 was also small in size, however, he had an average weight until approximately at the age of 11 years when he stopped steadily gaining weight and is currently at the 25th centile for weight while still maintaining a normal diet.
Recreational drug and alcohol usage during pregnancy was highly suspected; however, it could not be confirmed. The biological parents were unavailable during the clinical evaluation. Any possible role of drug or alcohol use during pregnancy in the siblings' phenotypes could not be established. Similarly, while both siblings presented with features of a connective tissue disorder with joint instability, scoliosis, and aortic root dilation, no DNA testing for these disorders were done during the initial clinical assessment. The known chromosome 1p31.1 deletion was the major focus of the clinical evaluation. Sibling 1 recently returned to our clinic, and genetic testing for a connective tissue disorder is under consideration. There is no information regarding a link between the NEGR1 gene and connective tissue disorders within the literature.
Learning disabilities and visual processing defects were considered findings in sibling 1 but not in sibling 2 who had no history of dyslexia or reading problems. After review of school performance and medical history forms, a definitive test for dyslexia was not performed in either sibling, therefore neither had a confirmed diagnosis of dyslexia. However, sibling 1 was given a visual performance profile, which indicated that he had oculomotor dysfunction and visual performance errors. Both siblings presented with ADHD and language impairments.
Evidence of dyslexia with errors in neuronal migration and connection in association with this chromosome region were noted in genome-wide screening and associated studies with a NEGR1 de novo CNV in 10 Indian families with dyslexia.14 Currently, there is a paucity of information regarding a link between ADHD or language impairment with this microdeletion, but intellectual disability and neurological impairment (e.g., abnormal muscle tone and strabismus) and central nervous system findings (e.g., microcephaly and brain anomalies) are recognized features. The authors encourage the reporting of additional subjects using chromosomal microarray analysis to identify subtle chromosome 1p31.1 deletions and the role of the NEGR1 gene to further characterize the phenotype and range of gene disturbances in this cytogenetic syndrome (Table 1).
Acknowledgments
We acknowledge support from the HD02528 grant from the National Institute of Child Health and Human Development.
References
- 1.Mircher C, Rethore M O, Lespinasse J, Fert-Ferrer S, Lundsteen C, Kirchoff M. Interstitial deletion of the short arm of chromosome 1: attempt to establish a clinical phenotype (46,XX,del (1)(p22p32)) Am J Med Genet A. 2003;118A(2):176–179. doi: 10.1002/ajmg.a.10052. [DOI] [PubMed] [Google Scholar]
- 2.Callier P, Faivre L, Thauvin-Robinet C. et al. Array-CGH in a series of 30 patients with mental retardation, dysmorphic features, and congenital malformations detected an interstitial 1p22.2-p31.1 deletion in a patient with features overlapping the Goldenhar syndrome. Am J Med Genet A. 2008;146A(16):2109–2115. doi: 10.1002/ajmg.a.32447. [DOI] [PubMed] [Google Scholar]
- 3.Chen C P, Su Y N, Chen Y Y. et al. Chromosome 1p32-p31 deletion syndrome: prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Taiwan J Obstet Gynecol. 2011;50(3):345–352. doi: 10.1016/j.tjog.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Maegawa G HB, Poplawski N K, Andresen B S. et al. Interstitial deletion of 1p22.2p31.1 and medium-chain acyl-CoA dehydrogenase deficiency in a patient with global developmental delay. Am J Med Genet A. 2008;146A(12):1581–1586. doi: 10.1002/ajmg.a.32255. [DOI] [PubMed] [Google Scholar]
- 5.Nava C, Keren B, Mignot C. et al. Prospective diagnostic analysis of copy number variants using SNP microarrays in individuals with autism spectrum disorders. Eur J Hum Genet. 2014;22(1):71–78. doi: 10.1038/ejhg.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yıldırım Y, Kerem M, Köroğlu Ç, Tolun A. A homozygous 237-kb deletion at 1p31 identified as the locus for midline cleft of the upper and lower lip in a consanguineous family. Eur J Hum Genet. 2014;22(3):333–337. doi: 10.1038/ejhg.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bene M, Duca-Marinescu A, Ioan D, Maximilian C. De novo interstitial deletion del(1)(p21p32) J Med Genet. 1979;16(4):323–327. doi: 10.1136/jmg.16.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen M B, Warburg M. Interstitial deletion 1p in a 30 year old woman. J Med Genet. 1987;24(4):229–231. doi: 10.1136/jmg.24.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai M M, Robards M F, Berry A C, Fear C N, Hart C. Two cases of interstitial deletion 1p. J Med Genet. 1991;28(2):128–130. doi: 10.1136/jmg.28.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flicek P, Amode M R, Barrell D. et al. Ensembl 2014. Nucleic Acids Res. 2014;42(Database issue):D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Funatsu N, Miyata S, Kumanogoh H. et al. Characterization of a novel rat brain glycosylphosphatidylinositol-anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J Biol Chem. 1999;274(12):8224–8230. doi: 10.1074/jbc.274.12.8224. [DOI] [PubMed] [Google Scholar]
- 12.Lee A W, Hengstler H, Schwald K. et al. Functional inactivation of the genome-wide association study obesity gene neuronal growth regulator 1 in mice causes a body mass phenotype. PLoS ONE. 2012;7(7):e41537. doi: 10.1371/journal.pone.0041537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boender A J, van Gestel M A, Garner K M, Luijendijk M C, Adan R A. The obesity-associated gene Negr1 regulates aspects of energy balance in rat hypothalamic areas. Physiol Rep. 2014;2(7):e12083. doi: 10.14814/phy2.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veerappa A M, Saldanha M, Padakannaya P, Ramachandra N B. Family-based genome-wide copy number scan identifies five new genes of dyslexia involved in dendritic spinal plasticity. J Hum Genet. 2013;58(8):539–547. doi: 10.1038/jhg.2013.47. [DOI] [PubMed] [Google Scholar]