

Abstract
Prolidase deficiency (PD) is a rare genetic disorder caused by mutations in the peptidase D (PEPD) gene, affecting collagen degradation. Features include lower extremity ulcers, facial dysmorphism, frequent respiratory infections, and intellectual disability, though there is significant intra- and interfamilial variability. Twenty-eight mutations have been previously reported, all either small deletions/duplications or point mutations discovered by enzyme or DNA assays. PD has been reported in patients of various ethnic backgrounds, but never in the Mexican-American population. We describe the first Mexican-American patient with PD, who presented with typical facial features, developmental delay, microcephaly, and xerosis. Chromosome microarray analysis (CMA) revealed a homozygous deletion in the region of 19q13.11, estimated to be between 124.79 and 195.72 kb in size, representing the largest PEPD gene deletion reported to date and the first discovered by CMA.
Keywords: Chromosome microarray analysis, Deletion mutation, Novel mutation, PEPD, Prolidase deficiency
Prolidase deficiency (PD; OMIM 170100) is a rare autosomal recessive inborn error of amino acid metabolism that affects collagen degradation, with an estimated incidence of 1-2 cases per 1,000,000 births [Lupi et al., 2008]. PD was first described by Goodman et al. [1968] and has a wide phenotypic spectrum with no evidence for genotype-phenotype correlation [Falik-Zaccai et al., 2010]. Common clinical features include facial dysmorphism, chronic ulcerations of the lower extremities, intellectual disability, and recurrent respiratory tract infections. The symptoms are caused by the reduction or loss of prolidase activity due to mutations in the peptidase D (PEPD; OMIM 613230) gene located in chromosome 19q13.11, which has multiple functions including involvement in the latter stages of catabolism of dietary and endogenous proteins rich in proline or hydroxyproline, such as collagen. Clinical manifestations are often detectable in early childhood, but late-onset cases have also been reported [Lupi et al., 2008].
Patient and Methods
Clinical Report
Our patient is a 6-year-old boy born to consanguineous parents (parents' fathers are second cousins) of Mexican descent. The remainder of the family history is unremarkable. Our patient presents with microcephaly (OFC = 48 cm, −8 SD) and with normal height (126 cm, 80th percentile) and weight (23 kg, 50th percentile). He has dysmorphic facial features including synophrys, proptosis, hypertelorism, downslanting palpebral fissures, a prominent nose, thin vermilion of the upper lip, prognathism, and low anterior and posterior hairlines. His skin examination revealed prominent supragluteal fat pads and diffuse xerosis. He has receptive and expressive language delay despite a normal hearing evaluation. He receives speech and occupational therapies, verbalizes ∼15 words and uses some sign language. He walked at the age of 19 months and currently walks and runs without difficulty. Other medical problems include constipation. Abdominal ultrasound revealed splenomegaly that was not detected on physical examination. His parents deny any history of ulcerations of the skin, recurrent pulmonary infections, or other recurrent infections.
Molecular Cytogenetic Analysis
Chromosome microarray analysis was performed using the array comparative genome hybridization (aCGH) method labeled with 2 different fluorophores for the patient extracted DNA and a sex-match control DNA and then hybridized onto NimbleGen CGX-12 v1.0 12-plex hg 18 based on the human genome build 18 following the manufacturer's protocol. The resolution of aberration is 10-50 kb for a detectable deletion of minimum size. The log2 ratio of the 2 different labeled patient and control DNA were analyzed and calculated using Genoglyphix database analysis (Signature, Spokane, Wash., USA).
Results
Molecular Cytogenetic Results
Based on the log2 ratio, the results revealed an interstitial biallelic deletion involving the PEPD gene (fig. 1). This deletion is characterized by a copy loss of 5 oligonucleotide probes in the region of 19q13.11. This abnormality is estimated to be a minimum size of 124.79 kb and a maximum size of 195.72 kb due to gaps in the regions represented on the microarray with genomic coordinates: arr[hg18] 19q13.11(38,569,683-38,704,639)×0. Because of the biallelic deletion, the patient has a homozygous deletion in PEPD, which is known to be associated with PD.
Fig. 1.

A aCGH Genoglyphix plot of chromosome 19 showing probes (blue dots) located at −3.0 log2 ratio. B UCSC plot showing the deletion involving all 15 exons and 14 introns of the PEPD gene.
FISH of the BAC clone RP11-1101H9 (205 kb with 62% within region of interest) directed onto 19q13.11 was obtained from Empire Genomics (Buffalo, N.Y., USA) and performed on the patient and his sister. FISH results showed 2 copies of diminished signals (compared to those found in the sister) in 50 interphase and 20 metaphase cells of the patient's peripheral blood (fig. 2B), indicating a biallelic deletion, confirming the aCGH findings. FISH of the same BAC clone performed on 50 interphase and 20 metaphase cells of the sister showed one diminished signal indicating a monoallelic deletion (fig. 2A, C).
Fig. 2.
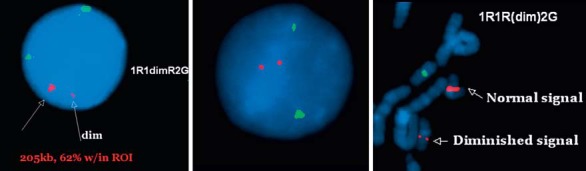
A Interphase cell of the sister showing 1 normal and 1 diminished signal (arrows) of the RP11-110H11 probe (SpectrumOrange™) in the presence of 2 normal signals of the centromere of chromosome 20 (SpectrumGreen™) used as an internal control. B Interphase cell of the patient showing 2 diminished signals of the probe. C Partial metaphase cell of the sister showing 1 diminished and 1 normal signal of the SpectrumOrange probe.
Biochemical Analysis
By ion exchange method, urine amino acid analysis revealed elevated methionine (195 mm/m creatinine; normal <50 mm/m creatinine) as well as unidentified peaks causing interference with the peaks of isoleucine, leucine and tyrosine. Using gas chromatography-mass spectrometry, urine organic acid analysis revealed imidodipeptiduria. Carbohydrate-deficient transferrin analysis was also performed to rule out a congenital disorder of glycosylation, given the physical finding of supragluteal fat pads. The result was normal.
Discussion
To date, ∼90 patients with PD have been described, 67 of which have been molecularly characterized revealing 28 distinct mutant alleles [Tanoue et al., 1990, 1991; Ledoux et al., 1994, 1996; Kikuchi et al., 2000; Forlino et al., 2002; Lupi et al., 2004, 2006, 2008; Hershkovitz et al., 2006; Wang et al., 2006; Falik-Zaccai et al., 2010; Klar et al., 2010; Butbul Aviel et al., 2012; Caselli et al., 2012; Pandit et al., 2013; Besio et al., 2015]. The PEPD gene is over 130 kb long and is comprised of 15 exons. Of the 28 mutations, there are 14 point mutations; 5 splice site mutations; 6 small deletions causing either a missing amino acid, a premature stop codon, or involving an intron; 1 larger deletion of 774 bp causing complete elimination of exon 14; 1 small duplication leading to a truncated enzyme, and one 3-bp insertion resulting in distortion of the enzyme (table 1). The current gene deletion is the largest yet reported. Additionally, all prior mutations were identified by enzyme or DNA assays, making this the first deletion identified by aCGH. The Databases of Decipher and International Standards for Cytogenomic Arrays have no patients with pure complete or intragenic PEPD deletions. All documented patients within 19q13.11 region have several genes including PEPD, and the Database of Genome Variants shows no variants.
Table 1.
List of molecularly characterized PD patients and their clinical manifestations
| Patient ID | Exon | Intron | Mutation | Effect on protein | Clinical phenotype |
Ethnicity/residence | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| skin ulcers | ID/DD | FI | FD | SM | additional features | |||||||
| 1a | 2 | 202–2A→T/202–2A→T | absent exon 3; alternative splicing | + | + | + | + | – | microcephaly, short stature, osteopenia, genu valgum | Turkey | Besio et al., 2015 | |
| 2a | 2 | 202–2A→T/202–2A→T | absent exon 3; alternative splicing | + | + | + | + | – | microcephaly, short stature, genu valgum | Turkey | Besio et al., 2015 | |
| 3 | 3 | 329+ 4de1/329+ 4del | + | – | – | + | – | microcephaly, short stature | Turkey | Besio et al., 2015 | ||
| 4 | 3 | 329+ 4de1/329+ 4del | + | + | + | + | – | microcephaly, osteopenia | Turkey | Besio et al., 2015 | ||
| 5 | 4 | IVS4– 1G→C/null allele | absent exon 5 | + | + | + | – | – | Oklahoma | Ledoux et al., 1994 | ||
| 6 | 6 | IVS6–2A→G/IVS6–2A→G | absent exon 7 | + | + | – | – | – | SLE | Canada | Ledoux et al., 1994 | |
| 7 | 7 | IVS7–1G→A/IVS7–ss1G→A | alternative splicing | + | + | + | + | + | anemia, telangiectasia, photosensitivity | Italy | Forlino et al., 2002 | |
| 8 | 7 | IVS7–1G→A/IVS7–1G→A | alternative splicing | + | – | + | + | – | telangiectasia | Italy | Forlino et al., 2002 | |
| 9 | 8 | 611duplAGGCCCACCGTGA/611duplAGGCCCACCGTGA | frameshift and premature stop codon | + | + | + | + | + | FTT | Turkey | Lupi et al., 2006 | |
| 10 | 8/12 | 551G→A/833G→A | Arg184Gln | – | – | – | – | – | asymptomatic at birth, no other data | Canada | Ledoux et al., 1996 | |
| 11 | 8 | 551C→T/551C→T | Arg184X | + | + | + | + | – | joint dislocations, partial deafness, visual disturbance | Japan | Kikuchi et al., 2000 | |
| 12 | 8 | 605C→T/605C→T | Ser202Phe | – | + | + | + | + | hepatomegaly, elevated IgE, brachydactyly, craniosynostosis | Druze/Israel | Hershkovitz et al., 2006 | |
| 13b | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | + | dermatitis | Moslem/Israel | Falik-Zaccai et al., 2010 | |
| 14b | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | – | Moslem/Israel | Falik-Zaccai et al., 2010 | ||
| 15b | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | – | Moslem/Israel | Falik-Zaccai et al., 2010 | ||
| 16b | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | – | Moslem/Israel | Falik-Zaccai et al., 2010 | ||
| 17b | 8 | 605C→T/605C→T | Ser202Phe | – | + | + | + | – | FTT, died at age 20 months of respiratory failure | Moslem/Israel | Falik-Zaccai et al., 2010 | |
| 18c | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | + | dermatitis | Moslem/Israel | Falik-Zaccai et al., 2010 | |
| 19c | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | + | dermatitis | Moslem/Israel | Falik-Zaccai et al., 2010 | |
| 20 | 8 | 605C→T/605C→T | Ser202Phe | – | – | + | – | + | hypotonia | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 21 | 8 | 605C→T/605C→T | Ser202Phe | – | + | + | + | – | dermatitis, asthma | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 22 | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | + | dermatitis, asthma | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 23 | 8 | 605C→T/605C→T | Ser202Phe | + | + | + | + | + | SLE phenotype | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 24d | 8 | 605C→T/605C→T | Ser202Phe | – | + | + | + | + | dermatitis, asthma, bronchiectasis | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 25d | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | – | asthma, dermatitis | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 26e | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | + | dermatitis | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 27e | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | – | FTT | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 28e | 8 | 605C→T/605C→T | Ser202Phe | – | + | – | + | + | cystic fibrosis, pseudo-psoriasis, FTT | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 29 | 8 | 605C→T/605C→T | Ser202Phe | – | – | – | + | – | SLE | Druze/Israel | Butbul Aviel et al., 2012 | |
| 30 | 8 | 605C→T/605C→T | Ser202Phe | – | + | + | + | + | SLE, pancytopenia, lung disease, rash | Druze/Israel | Butbul Aviel et al., 2012 | |
| 31 | 9 | 634G→C/634G→C | Ala212Pro | – | + | + | + | + | SLE, FTT, microcephaly, clinodactyly, brachydactyly | Moslem Arab | Butbul Aviel et al., 2012 | |
| 32 | 9 | 634G→C/634G→C | Ala212Pro | – | + | + | + | + | FTT, dermatitis, SLE phenotype | Moslem/Israel | Falik-Zaccai et al., 2010 | |
| 33 | 10 | 691delTAC/691delTAC | 231delTyr | + | + | + | + | – | hypotonia, irritability | Portugal | Lupi et al., 2004; Pereira and Vilarinho, 1997 | |
| 34 | 10 | 691delTAC/691delTAC | 231delTyr | + | – | + | + | – | elevated IgE | Portugal | Lupi et al., 2004; | |
| Lopes et al., 2002 | ||||||||||||
| 35f | 11 | 768C→G/768C→G | Tyr256X | + | – | – | + | + | photosensitivity, elevated IgE, SLE phenotype | Moslem Arab | Klar et al., 2010 | |
| 36f | 11 | 768C→G/768C→G | Tyr256X | + | – | + | + | + | telangiectasia, SLE phenotype | Moslem Arab | Klar et al., 2010 | |
| 37f | 11 | 768C→G/768C→G | Tyr256X | + | – | + | – | + | fulminant hepatitis leading to infant death | Moslem Arab | Klar et al., 2010 | |
| 38 | 11 | IVS11 + 1G→C/null allele | absent exon 11 | + | + | – | + | – | telangiectasia, photosensitivity | Italy | Forlino et al., 2002 | |
| 39 | 11 | IVS11 + 1G→A/ IVS11 + 1G→A | alternative splicing | – | – | + | – | – | India | Lupi et al., 2008 | ||
| 40g | 11 | 793C→T/793C→T | Arg265X | + | + | + | + | – | hepatomegaly, eczema-like rash, asplenia, FTT | Amish/Ohio | Wang et al., 2006 | |
| 41g | 11 | 793C→T/793C→T | Arg265X | + | + | + | + | + | hepatomegaly, eczema-like rash, FTT | Amish/Ohio | Wang et al., 2006 | |
| 42g | 11 | 793C→T/793C→Τ | Arg265X | + | – | + | + | + | hepatomegaly, eczema-like rash | Amish/Ohio | Wang et ah, 2006 | |
| 43g | 11 | 793C→T/793C→Τ | Arg265X | + | – | + | + | + | hepatomegaly, eczema-like rash | Amish/Ohio | Wang et al., 2006 | |
| 44 | 12 | 826G→A/826G→A | Asp276Asn | + | – | – | – | + | Middle East | Tanoue et al., 1990; | ||
| Lapière and Nusgens, 1969 | ||||||||||||
| 45 | 12 | 826G→A/826G→A | Asp276Asn | + | + | + | - | - | hypotonia, eczema-like rash, joint laxity, cortical and cerebellar atrophy on MRI brain | Chinese | Tanoue et al., 1990; Lombeck et al., 1986 | |
| 46h | 12 | 909_910insGCC | Q303_K304insA | + | - | - | - | - | Kashmir | Pandit et al., 2013 | ||
| 47h | 12 | 909_910insGCC | Q303_K304insA | + | – | – | – | – | Kashmir | Pandit et al., 2013 | ||
| 48 | 12 | 874delT/874delT | premature stop codon | + | – | + | – | – | osteopenia | Turkey | Besio et al., 2015 | |
| 49 | 12 | 977G→A/977G→A | Trp325* | + | + | – | + | – | genu valgum | Spain | Besio et al., 2015 | |
| 50 | 12/14 | 977G→A/1342G→A | Trp325*/Gly448Arg | + | + | + | + | – | short stature, genu valgum | England | Besio et al., 2015 | |
| 51 | 12/14 | 826G→A/1342G→A | Asp276Asn/Gly448Arg | + | + | – | – | – | perianal eczema | Denmark | Lupi et al., 2006 | |
| 52 | 12/14 | 833G→A/1342G→A | Gly278Asp/Gly448Arg | + | + | + | – | + | Denmark | Lupi et al., 2006 | ||
| 53i | 13 | 1103T→G/1103T→G | Leu368Arg | – | + | – | + | + | FTT | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 54i | 13 | 1103T→G/1103T→G | Leu368Arg | – | + | – | + | + | FTT | Druze/Israel | Falik-Zaccai et al., 2010 | |
| 55 | 13/14 | 1133delACG/1301delT | Asp378del/frameshift and | + | + | + | + | + | short stature, microcephaly | Italy | Caselli et al., 2011 | |
| 56j | 14 | 1234G→A/1234G→A | premature stop codon Glu412Lys | + | – | – | + | – | elevated IgE, hemoglobin O trait, eczema-like rash, dry skin | Turkey | Lupi et al., 2006; Aytug et al., 2006 | |
| 57j | 14 | 1234G→A/1234G→A | Glu412Lys | – | – | – | – | – | slow skin healing after injury | Turkey | Lupi et al., 2006 | |
| 58 | 14 | 1342G→A/null allele | Gly448Arg | + | + | + | – | – | Canada | Ledoux et al., 1994 | ||
| 59 | 14 | 1342G→A/1342G→A | Gly448Arg | + | + | + | + | + | UK | Ledoux et al., 1994 | ||
| 60k | 14 | 1342G→A/1342G→A | Gly448Arg | + | + | + | + | – | Italy | Forlino et al., 2002 | ||
| 61k | 14 | 1342G→A/1342G→A | Gly448Arg | + | – | – | – | – | Italy | Forlino et al., 2002 | ||
| 62 | 14 | 1342G→A/1342G→A | Gly448Arg | + | + | – | – | + | SLE | Italy | Lupi et al., 2006 | |
| 63l | 14/15 | 1342G→A/1354G→A | Gly448Arg/Glu451 Lys | + | + | + | + | – | osteopenia | Poland | Besio et al., 2015 | |
| 64l | 14/15 | 1342G→A/1354G→A | Gly448Arg/Glu451 Lys | + | – | + | + | – | osteopenia | Poland | Besio et al., 2015 | |
| 65m | 14 | del774bp | absent exon 14 | + | + | – | – | – | telangiectasia, premature gray hairs at age 13 years | Japan | Tanoue et al., 1991; Arata et al., 1979 | |
| 66m | 14 | del774bp | absent exon 14 | + | – | – | – | – | Japan | Tanoue et al., 1991; | ||
| Arata et al., 1979 | ||||||||||||
| 67 | 15 | 1354delGAG/null allele | 452delGlu | + | + | chronic liver disease | Australia | Ledoux et al., 1994 | ||||
| 68 | 124.79–195.72-kb deletion | – | – | – | + | – | microcephaly, lipodystrophy, xerosis | Mexican/USA | Current paper | |||
Table adapted from Lupi et al. [2008] with additional information on mutations and phenotypes. Patients are unrelated unless otherwise indicated. DD = Developmental delay; FD = facial dysmorphism; FI = frequent infections; FTT = failure to thrive; ID = intellectual disability; SM = splenomegaly.
Siblings;
family related;
sisters;
cousins;
brothers.
Prolidase is a dipeptidase necessary for the final step of collagen degradation. It splits imidodipeptides with C-terminal proline or hydroxyproline, making proline available for reutilization in collagen synthesis [Myara et al., 1984; Klar et al., 2010]. In PD, this enzyme has decreased or complete loss of activity, giving rise to imidodipeptiduria and a variety of clinical manifestations. In our patient, the urine amino acids showed ‘interfering peaks’ which are consistent with prolidase peptide deficiency. The urine organic acids were able to identify imidodipeptides.
PD exhibits significant inter- and intrafamilial phenotypic variability [Falik-Zaccai et al., 2010]. Among patients with molecular characterization, skin abnormalities are the most common feature, present in 79% of patients, including ulcerations, telangiectasias, photosensitivity, dry skin, pseudo-psoriasis, or nonspecific dermatitis. Other features include dysmorphism (71%), intellectual disability or developmental delay (69%), frequent or serious infections (53%), splenomegaly (41%), bony abnormalities (19%), systemic lupus erythematosus (SLE; 14%), failure to thrive (16%), microcephaly (12%), hepatomegaly (9%), and asthma (7%), as outlined in table 1. Interestingly, our patient did not have a history of frequent infections or skin ulcerations despite the large deletion. Additionally, our patient has a unique finding, supragluteal lipodystrophy, which has not been previously reported in PD. For this reason, the carbohydrate-deficient transferrin studies were tested and found negative, ruling out congenital disorders of glycosylation.
Notably, SLE has been linked to PD [Bissonnette et al., 1993; Shrinath et al., 1997; Di Rocco et al., 2007] and is associated with multiple mutations [Lupi et al., 2006, 2008; Falik-Zaccai et al., 2010; Klar et al., 2010; Butbul Aviel et al., 2012]. The mechanistic relationship between these entities is not clear, but a loss of immune tolerance to lupus-associated autoantigens in PD has been hypothesized, leading to positive antinuclear antibodies and other autoantibody testing even in patients without clinical SLE [Kurien et al., 2013].
Cystic fibrosis confirmed by sweat chloride testing has also been found in patients with PD, though mutation and haplotype analysis did not show changes in the CFTR gene [Luder et al., 2007]. Multiple other PD patients with clinical cystic fibrosis have had normal sweat chloride testing [Mandel et al., 2000]. Despite nullisomic complete deletion in our patient, SLE or cystic fibrosis features were not observed. This suggests that the presence of a mutated protein may cause more systemic symptoms when compared to an absent protein. However, more patients with a similar size deletion would be needed to confirm this. It is also possible that SLE or chronic lung disease have not yet developed in our patient, as other cases appear to present in adolescence and adulthood.
The diagnosis in our patient was made incidentally at the age of 4 years via aCGH that was ordered due to dysmorphic features, microcephaly and developmental delay. If a mutation had not been detected, the diagnosis may not have been made, given his lack of several classically described symptoms. However, our patient does have the 3 most common features found in molecularly characterized patients: dysmorphism, developmental delay and abnormal skin findings. Therefore, we suggest that PD should be part of the differential diagnosis for any patient with characteristic facial dysmorphism, abnormal skin findings of any kind, and intellectual disability or developmental delay as they are each present in over 50% of patients.
Our patient's sister, found to be heterozygous for the same deletion, is asymptomatic. The patient's parents are obligate carriers and are also asymptomatic. Thus, there is no evidence of haploinsufficiency of the gene, consistent with previous reports of families with multiple carriers [Falik-Zaccai et al., 2010].
The ethnic origin for previously reported patients with PD is in European countries, Canada, the US, and Middle Eastern and several Asian countries (table 1). To our knowledge, there have been no reported cases in patients of Mexican descent, making our patient the first.
In conclusion, we report a novel large deletion of the PEPD gene, discovered by aCGH in a Mexican-American family. Although PD is a rare disorder, it is likely underdiagnosed, particularly in patients with a mild phenotype. The marked variability in clinical presentation also creates diagnostic difficulty, and the prognosis is unpredictable. For this reason, genetic counseling, even to members of the same family, is problematic [Mandel et al., 2000]. No specific therapies can be recommended for PD. Greater awareness and improved understanding of the pathophysiology of the disease will improve the recognition and treatment of this multisystem disorder.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have no conflicts of interest to disclose.
References
- 1.Arata J, Umemura S, Yamamoto Y, Hagiyama M, Nohara N. Prolidase deficiency, its dermatological manifestation and some additional biochemical studies. Arch Dermatol. 1979;115:62–67. doi: 10.1001/archderm.115.1.62. [DOI] [PubMed] [Google Scholar]
- 2.Aytug AF, Ergun T, Ratip S, Elcioglu N, Gultepe M, et al. Prolidase deficiency associated with hemoglobin O trait and microcytic anemia. Int J Dermatol. 2006;45:867–868. doi: 10.1111/j.1365-4632.2006.02690.x. [DOI] [PubMed] [Google Scholar]
- 3.Besio R, Maruelli S, Gioia R, Villa I, Grabowski P, et al. Lack of prolidase causes a bone phenotype both in human and in mouse. Bone. 2015;72:53–64. doi: 10.1016/j.bone.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Bissonnette R, Friedmann D, Giroux JM, Dolenga M, Hechtman P, et al. Prolidase deficiency: a multisystemic hereditary disorder. J Am Acad Dermatol. 1993;29:818–821. doi: 10.1016/0190-9622(93)70245-o. [DOI] [PubMed] [Google Scholar]
- 5.Butbul Aviel Y, Mandel H, Hersh EA, Bergman R, Adiv OE, et al. Prolidase deficiency associated with systemic lupus erythematosus (SLE): single site experience and literature review. Pediatr Rheumatol Online J. 2012;10:18. doi: 10.1186/1546-0096-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caselli D, Cimaz R, Besio R, Rossi A, De Lorenzi E, et al. Partial rescue of biochemical parameters after hematopoietic stem cell transplantation in a patient with prolidase deficiency due to two novel PEPD mutations. JIMD Rep. 2012;3:71–77. doi: 10.1007/8904_2011_62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Rocco M, Fantasia AR, Taro M, Loy A, Forlino A, Martini A. Systemic lupus erythematosus-like disease in a 6-year-old boy with prolidase deficiency. J Inherit Metab Dis. 2007;30:814. doi: 10.1007/s10545-007-0496-z. [DOI] [PubMed] [Google Scholar]
- 8.Falik-Zaccai TC, Khayat M, Luder A, Frenkel P, Magen D, et al. A broad spectrum of developmental delay in a large cohort of prolidase deficiency patients demonstrates marked interfamilial and intrafamilial phenotypic variability. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:46–56. doi: 10.1002/ajmg.b.30945. [DOI] [PubMed] [Google Scholar]
- 9.Forlino A, Lupi A, Vaghi P, Icaro Cornaglia A, Calligaro A, et al. Mutation analysis of five new patients affected by prolidase deficiency: the lack of enzyme activity causes necrosis-like cell death in cultured fibroblasts. Hum Genet. 2002;111:314–322. doi: 10.1007/s00439-002-0792-5. [DOI] [PubMed] [Google Scholar]
- 10.Goodman SI, Solomons CC, Muschenheim F, McIntyre CA, Miles B, O'Brien DL. A syndrome resembling lathyrism associated with iminodipeptiduria. Am J Med. 1968;45:152–159. doi: 10.1016/0002-9343(68)90016-8. [DOI] [PubMed] [Google Scholar]
- 11.Hershkovitz T, Hassoun G, Indelman M, Shlush LI, Bergman R, et al. A homozygous missense mutation in PEPD encoding peptidase D causes prolidase deficiency associated with hyper-IgE syndrome. Clin Exp Dermatol. 2006;31:435–440. doi: 10.1111/j.1365-2230.2006.02112.x. [DOI] [PubMed] [Google Scholar]
- 12.Kikuchi S, Tanoue A, Endo F, Wakasugi S, Matsuo N, Tsujimoto G. A novel nonsense mutation of the PEPD gene in a Japanese patient with prolidase deficiency. J Hum Genet. 2000;45:102–104. doi: 10.1007/s100380050023. [DOI] [PubMed] [Google Scholar]
- 13.Klar A, Navon-Elkan P, Rubinow A, Branski D, Hurvitz H, et al. Prolidase deficiency: it looks like systemic lupus erythematosus but it is not. Eur J Pediatr. 2010;169:727–732. doi: 10.1007/s00431-009-1102-1. [DOI] [PubMed] [Google Scholar]
- 14.Kurien BT, D'Sousa A, Bruner BF, Gross T, James JA, et al. Prolidase deficiency breaks tolerance to lupus-associated antigens. Int J Rheum Dis. 2013;16:674–680. doi: 10.1111/1756-185X.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lapiére CM, Nusgens B. Torpid skin wounds and collagen metabolism disorder (in French) Arch Belg Dermatol Syphiligr. 1969;25:353–356. [PubMed] [Google Scholar]
- 16.Ledoux P, Scriver C, Hechtman P. Four novel PEPD alleles causing prolidase deficiency. Am J Hum Genet. 1994;54:1014–1021. [PMC free article] [PubMed] [Google Scholar]
- 17.Ledoux P, Scriver CR, Hechtman P. Expression and molecular analysis of mutations in prolidase deficiency. Am J Hum Genet. 1996;59:1035–1039. [PMC free article] [PubMed] [Google Scholar]
- 18.Lombeck I, Wendel U, Versieck J, van Ballenberghe L, Bremer HJ, et al. Increased manganese content and reduced arginase activity in erythrocytes of a patient with prolidase deficiency (iminodipeptiduria) Eur J Pediatr. 1986;144:571–573. doi: 10.1007/BF00496038. [DOI] [PubMed] [Google Scholar]
- 19.Lopes I, Marques L, Neves E, Silva A, Taveira M, et al. Prolidase deficiency with hyperimmunoglobulin E: a case report. Pediatr Allergy Immunol. 2002;13:140–142. doi: 10.1034/j.1399-3038.2002.00075.x. [DOI] [PubMed] [Google Scholar]
- 20.Luder AS, Mandel H, Khayat M, Gurevich I, Frankel P, et al. Chronic lung disease and cystic fibrosis phenotype in prolidase deficiency: a newly recognized association. J Pediatr. 2007;150:656–658. doi: 10.1016/j.jpeds.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Lupi A, De Riso A, Torre SD, Rossi A, Campari E, et al. Characterization of a new PEPD allele causing prolidase deficiency in two unrelated patients: natural-occurrent mutations as a tool to investigate structure-function relationship. J Hum Genet. 2004;49:500–506. doi: 10.1007/s10038-004-0180-1. [DOI] [PubMed] [Google Scholar]
- 22.Lupi A, Rossi A, Campari E, Pecora F, Lund AM, et al. Molecular characterisation of six patients with prolidase deficiency: identification of the first small duplication in the prolidase gene and of a mutation generating symptomatic and asymptomatic outcomes within the same family. J Med Genet. 2006;43:e58. doi: 10.1136/jmg.2006.043315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lupi A, Tenni R, Rossi A, Cetta G, Forlino A. Human prolidase and prolidase deficiency: an overview on the characterization of the enzyme involved in proline recycling and on the effects of its mutations. Amino Acids. 2008;35:739–752. doi: 10.1007/s00726-008-0055-4. [DOI] [PubMed] [Google Scholar]
- 24.Mandel H, Abeling N, Gutman A, Berant M, Scholten EG, et al. Prolidase deficiency among an Israeli population: prenatal diagnosis in a genetic disorder with uncertain prognosis. Prenat Diagn. 2000;20:927–929. doi: 10.1002/1097-0223(200011)20:11<927::aid-pd943>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 25.Myara I, Charpentier C, Lemonnier A. Prolidase and prolidase deficiency. Life Sci. 1984;34:1985–1998. doi: 10.1016/0024-3205(84)90363-1. [DOI] [PubMed] [Google Scholar]
- 26.Pandit RA, Chen CJ, Butt TA, Islam N. Identification and analysis of a novel mutation in PEPD gene in two Kashmiri siblings with prolidase enzyme deficiency. Gene. 2013;516:316–319. doi: 10.1016/j.gene.2012.12.070. [DOI] [PubMed] [Google Scholar]
- 27.Pereira JS, Vilarinho L. Doença metabólica rara. Deficiência em prolidase. Acta Pediatr Port. 1997;28:237–239. [Google Scholar]
- 28.Shrinath M, Walter JII, Haeney M, Couriel JM, Lewis MA, Herrick AL. Prolidase deficiency and systemic lupus erythematosus. Arch Dis Child. 1997;76:441–444. doi: 10.1136/adc.76.5.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanoue A, Endo F, Kitano A, Matsuda I. A single nucleotide change in the prolidase gene in fibroblasts from two patients with polypeptide positive prolidase deficiency. Expression of the mutant enzyme in NIH 3T3 cells. J Clin Invest. 1990;86:351–355. doi: 10.1172/JCI114708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanoue A, Endo F, Akaboshi I, Oono T, Arata J, Matsuda I. Molecular defect in siblings with prolidase deficiency and absence or presence of clinical symptoms. A 0.8-kb deletion with breakpoints at the short, direct repeat in the PEPD gene and synthesis of abnormal messenger RNA and inactive polypeptide. J Clin Invest. 1991;87:1171–1176. doi: 10.1172/JCI115115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Kurien BT, Lundgren D, Patel NC, Kaufman KM, et al. A nonsense mutation of PEPD in four Amish children with prolidase deficiency. Am J Med Genet A. 2006;140:580–585. doi: 10.1002/ajmg.a.31134. [DOI] [PubMed] [Google Scholar]